

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Familial dysalbuminemic hyperthyroxinemia (FDH) is the most common inherited cause of high serum total thyroxine (T4) concentrations in clinically euthyroid subjects, showing an autosomal dominant transmission with an estimated prevalence of 1 in 10,000 [1]. Spurious TFT may lead to inappropriate treatments including medical or ablative therapy [2].
Variants at codons 90, 242, and 246 (conventional codons 66, 218, and 222, respectively) in the albumin gene (ALB) have been reported to be underlying causes of FDH, mainly by producing an abnormal human serum albumin (HSA) with enhanced T4 binding affinity. In 1994, two independent groups described a p.Arg242His variant in Caucasian individuals [34] followed by reports in Hispanic/Puerto Rican [5] and Chinese subjects [6]. In 1997, Wada et al [7] reported a p.Arg242Pro variant in Japanese cases, showing 80-fold increased T4 binding affinity, and subsequent cases were described in a Swiss family [8]. In 1998, another ALB variant (p.Leu90Pro) producing an HSA with increased affinity for T3 was found in the Thai population [9]. Recently, a novel variant in codon 242 (p.Arg242Ser) [10] and another variant in codon 246 (p.Arg246Ile) have also been reported [11].
Although FDH might not be rare in the Korean population, there have been no confirmed cases in Korea. We recently found a Korean family with FDH. The proband (II-2) was a 38-yr-old Korean woman. Five years ago, she was transferred to a tertiary hospital owing to abnormal TFT results; however, her thyroid function was normal when examined by a different assay. Resistance to thyroid hormone (RTH) was excluded with a negative result upon thyroid hormone receptor β gene (THRB) analysis.
This year, she underwent a routine check-up for TFT and had a high FT4 of 2.55 ng/dL (reference range 0.95-1.57 ng/dL) with normal TSH as determined by using the Roche e601 Cobas 6000 (Roche Diagnostics, West Sussex, England). Then she was transferred to our institution, and her blood sample was tested with two different TFT assays (Table 1).
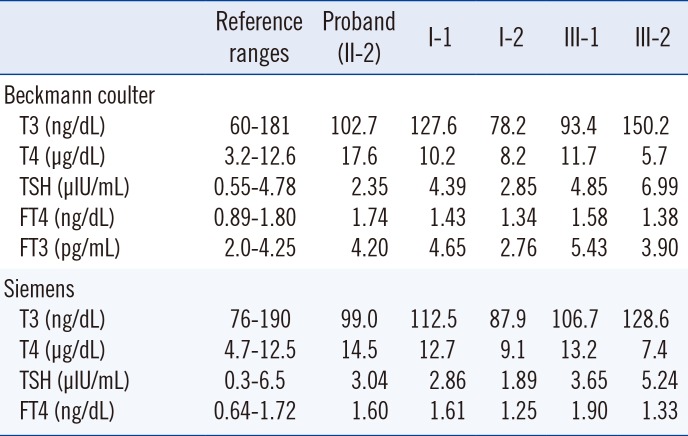
Table 1
Thyroid function tests for the proband and family members with familial dysalbuminemic hyperthyroxinemia
![]()
Using the Beckman Coulter RIA kit (Beckman Coulter, Brea, CA, USA), TFT results were as follows: 102.7 ng/dL for T3 (reference range 60-181 ng/dL), 17.6 µg/dL for T4 (reference range 3.2-12.6 µg/dL), 2.35 µIU/mL TSH (reference range 0.55-4.78 µIU/mL), 1.74 ng/dL FT4 (reference range 0.89-1.80 ng/dL), and 4.20 pg/mL FT3 (reference range 2.0-4.25 ng/dL). With the Siemens AG ADVIA Centaur kit (ADVIA Centaur AG, Munich, Germany), TFT values were as follows: 99 ng/dL T3 (reference range 76-190 ng/dL), 14.5 µg/dL T4 (reference range 4.7-12.5 µg/dL), 3.04 µIU/mL TSH (reference range 0.3-6.5 µIU/mL), and 1.60 ng/dL FT4 (reference range 0.64-1.72 ng/dL). A repeated test of the same blood sample with the Roche e601 Cobas 6000 showed a high FT4 (2.49-2.68 ng/dL, reference range 0.95-1.57 ng/dL).
Among first-degree relatives tested (I-1, I-2, III-1, and III-2; Fig. 1), the proband's father (I-1) and eldest son (III-1) showed similar TFT patterns with slightly elevated T4 (12.7 and 13.2 µg/dL, respectively with the Siemens AG ADVIA Centaur kit), but normal TSH and T3. The eldest son had a high FT4 of 1.90 ng/dL using the Siemens AG ADVIA Centaur kit. Nevertheless, two individuals were in a clinically euthyroid state.
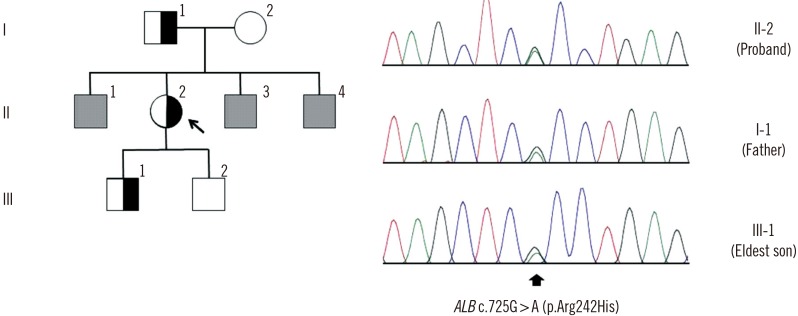 | Fig. 1Pedigree of the familial dysalbuminemic hyperthyroxinemia (FDH) family and albumin gene (ALB) mutation analysis for the proband. Half-closed and open squares indicate male subjects with and without FDH, respectively. Half-closed and open circles represent female subjects with and without FDH, respectively. Grey squares indicate male subjects who were not tested for thyroid function or ALB mutation. The arrow indicates the proband. Sequencing analysis identified the c.725G>A (p.Arg242His) mutation (arrow).
|
In order to confirm FDH, ABL was analyzed by using Sanger sequencing. After obtaining informed consent, genomic DNA was extracted from peripheral blood leukocytes by using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. Considering that the p.Arg242 residue is a hotspot in FDH, exon 7 of the ABL containing this residue was PCR-amplified and sequenced on an ABI 3730xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) using a BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems), and the sequencing results revealed the c.725G>A (p.Arg242His) variant in the proband (Fig. 1).
This variant was not present in data from 622 Korean control exomes or approximately 8,600 East Asian alleles from the Exome Aggregation Consortium (ExAC). The proband's father and eldest son with elevated T4 were also heterozygous for this variant; however, the proband's mother and younger son who had normal TFT results did not have this variant.
The p.Arg242His variant results in a mild form of FDH that produces HSA with enhanced affinity for T4 (10- to 15-fold increase) and results in high T4 levels with a 2-fold increase [346]. In our report, the proband had T4 levels that were increased 1.2- to 1.4-fold, which exceeded the upper reference limit. Two affected family members (I-1, III-1) also had slightly elevated T4 values. Using the two kits from Beckmann Coulter and Siemens, FT4 levels were normal in the proband, although the concentrations were close to upper normal limits in each assay. Notably, her FT4 level was above the reference interval using the Roche kit, even with the same blood sample.
Different FT4 assays have considerable variation in measurements. Generally, 2-step assays, in which a washing step immediately after capture prevents the T4 analog and serum albumin from coming into contact, are expected to yield normal FT4 levels in FDH patients. However, according to a study by Cartwright et al [12] that examined free T4 values in FDH individuals using different assays, including 1- and 2-step methodologies, some 2-step assays yielded spuriously high values in individuals with this condition. The Siemens Centaur 1-step assay performed considerably better than some of the other 2-step methods. This result is likely because inhibitors of T4 and T4 analogs bind to the albumin, and the chloride concentration in the incubation buffer plays a role [13].
Accurate FDH diagnosis is important because the affected individual can undergo unnecessary evaluation and treatment due to misdiagnosis. T4 concentrations vary according to the foci of FDH variants, and the results of FT4 are unreliable between different assays; therefore, clinical suspicion is critical. We believe that FDH cases are underestimated and underdiagnosed owing to insufficient clinical suspicion. Clinicians should consider the possibility of FDH in subjects with a clinically euthyroid state, but with abnormal TFT results.
 XML Download
XML Download