

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Antley-Bixler syndrome (ABS) is a rare congenital multiple mal formation syndrome caused by mutations in fibroblast growth factor receptor 2 (FGFR2) and cytochrome p450 oxidoreduc tase (POR) genes [123]. FGFR2-related ABS is autosomal domi nant, while POR-related ABS is autosomal recessive. In addition to skeletal malformations, POR mutation also causes glucocorti-coid deficiency and congenital adrenal hyperplasia with ambig uous genitalia in both sexes [45].
FGFR2 encodes a transmembrane receptor tyrosine kinase, and POR encodes cytochrome p450 oxidoreductase, which transfers electrons to microsomal enzymes, including three ste roidogenic enzymes: P450c17 (17α-hydroxylase/17,20 lyase), P450c21 (21-hydroxylase), and P450aro (aromatase) [6].
In Korea, one case of FGFR2-related ABS and two cases of POR-related ABS have been reported thus far [78]. Here we present a patient with ABS carrying a novel likely pathogenic vari ant in the POR gene.
A 21-yr-old woman with delayed puberty, loss of pubic hair, and skeletal anomalies presented to the Department of Medi-cine, Heart Vascular Stroke Institute of Samsung Medical Cen ter, Seoul, Korea. She was of normal stature and development, compared with general Korean standards. Her prenatal history was unremarkable. Mild facial dysmorphism was noted: micro gnathia, high arched palate, and low-set deformed ears. Multi ple skeletal abnormalities, including shortening of the fourth metatarsal bones, delayed closure of the hand and foot growth plates, and bilateral elbow dysplasia, were observed (Fig. 1). Ul trasonography revealed an underdeveloped uterus. Although baseline 17-α-OH-progesterone level was elevated to 531 ng/dL, adrenocorticotropic hormone (ACTH) was within the normal range (27.1 pg/mL). In the rapid ACTH test, cortisol level de creased from 14.6 µg/dL to 13.1 µg/dL, and aldosterone level increased from 71.0 ng/dL to 102.7 ng/dL. On the basis of the clinical signs of skeletal anomaly and secondary amenorrhea, the patient was suspected of having ABS. However, compared with previous reports, this patient had a relatively weak phenotype with respect to impaired sexual development and steroido genesis. We performed exome sequencing instead of targeted Sanger sequencing for the following reasons: (1) the patient had a mild phenotype; (2) the FGFR2 and POR genes contain a large number of exons, and conventional gene-by-gene sequencing would be more expensive and time-consuming than exome se quencing, and (3) exome sequencing would allow for the analy sis of new ABS candidate genes.
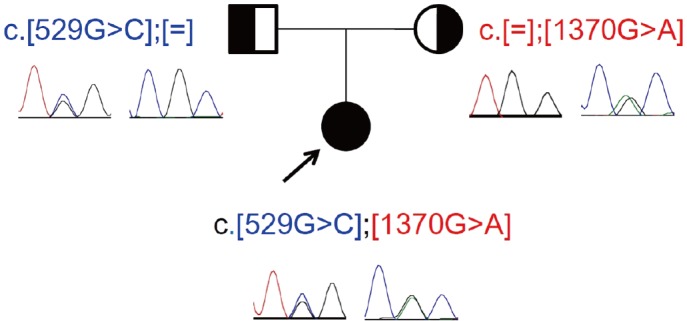
After obtaining informed consent, genomic DNA was extracted and purified by using the Agilent SureSelect Human All Exon v5 Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a HiSeq 2000 platform (Illumina, Inc., San Diego, CA, USA). By exome sequencing, a known POR pathogenic variant, NM_000941.2: c.1370G>A (p.Arg457His) and a novel POR mis sense variant, c.529G>C expected to cause an amino acid sub stitution (p.Gly177Arg), were identified in the proband. There were no suspected pathogenic variants in the FGFR2 gene. Sanger se quencing validated the c.529G>C (p.Gly177Arg) and c.1370G>A (p.Arg457His) variants in the POR gene. Analysis of her parents confirmed that the c.529G>C (p.Gly177Arg) variant was inher ited from the father, and c.1370G>A (p.Arg457His) from the mother (Fig. 2).
The c.1370G>A (p.Arg457His) variant has been reported in Korean patients with ABS, and is a known global founder muta tion causing ABS [789]. The p.Gly177Arg variant was absent from the single nucleotide polymorphism database (dbSNP) (build 149), the Exome Aggregation Consortium (http://exac.broadinstitute.org/), and the Korean reference genome data bases. In silico analysis, with both SIFT (http://sift.bii.a-star.edu.sg/index.html) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), predicted p.Gly177Arg to be deleterious. Thus, we conclude that this variant is likely pathogenic [10].
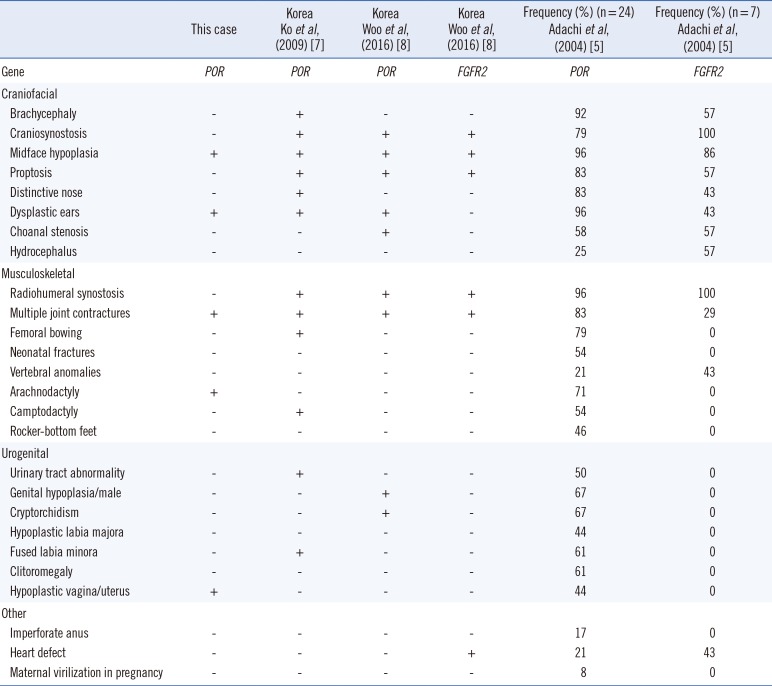
POR-related ABS is usually diagnosed at a younger age relative to FGFR2-related ABS, because it causes congenital adrenal hyperplasia. However, the present case had a relatively weak phe notype and was diagnosed over the age of 20 years. Neverthe less, the clinical features of this patient were similar to those re ported for POR mutations relative to FGFR2 mutations (Table 1).
In conclusion, we have identified a novel, likely pathogenic variant (c.529G>C; p.Gly177Arg) in the POR gene and this is the third reported case of ABS in Korea. This report will contrib ute to a better understanding of the genetic background of Ko rean patients with ABS.

 XML Download
XML Download