

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Stargardt-like macular dystrophy 4 (STGD4; OMIM No. 603786), an extremely rare autosomal dominant macular dystrophy, is characterized by bilateral bull's eye atrophy of the macula and the presence of yellow flecks in the posterior pole [12]. Patients with STGD4 show decreased central vision during the first several decades of life, which often progresses to severe vision loss with visual acuity of 20/200 or less [1234]. STGD4 is caused by mutations in the PROM1 gene (encoding prominin-1) on chromosome 4 [2]. The p.R373C missense mutation in the PROM1 gene causes several types of autosomal dominant retinal degeneration, including STGD4, bull's eye macular dystrophy (MCDR2), retinitis pigmentosa, and cone-rod dystrophy [23]. It has been proposed that prominin-1, a 5-transmembrane glycoprotein also known as CD133, is involved in photoreceptor disk morphogenesis [2]. The dysfunction of this protein may cause serious visual impairment. We report the first case of a PROM1 p.R373C mutation in a Korean patient with an STGD4 phenotype, using an exome-sequencing panel of known inherited retinal degeneration genes.
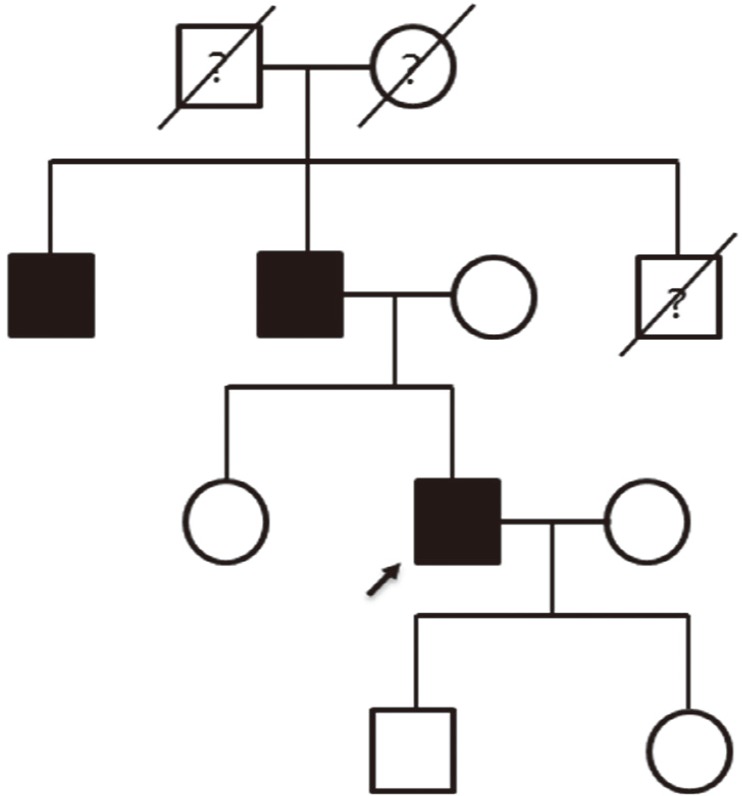
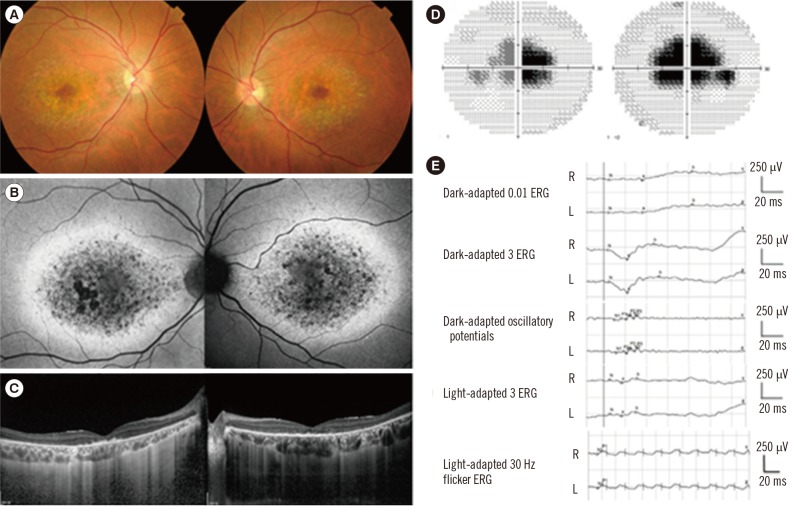
This study was approved by the Institutional Review Board at Samsung Medical Center (IRB No. 2014-09-052), Seoul, Korea, and written informed consent was obtained from the patient. A 38-yr old man visited the Retina Clinic at Samsung Medical Center complaining of central visual disturbance without photophobia. He has a family history of visual impairment, suggesting an autosomal dominant inheritance pattern (Fig. 1). A review of systems revealed no remarkable findings. His decimal best corrected visual acuity was 0.6 in both eyes. Slit lamp biomicroscopic examination showed no remarkable findings in anterior segments. Fundus examination showed bilateral atrophic macular lesions with small flecks in the posterior pole (Fig. 2A). Fundus autofluorescence showed a bull's-eye pattern of hypo-autofluorescent macular lesions surrounded by hyper-autofluorescence (Fig. 2B). Spectral-domain optical coherence tomography (Spectralis; Heidelberg Engineering, Heidelberg, Germany) showed retinal pigment epithelium atrophy and photoreceptor layer defects (Fig. 2C). An automated visual field test (Humphrey C30-2) showed bilateral central scotoma (Fig. 2D). Full-field electroretinogram (ERG, Ganzfeld Q450; Roland Consult, Brandenburg a.d. Havel, Germany) according to the International Society for Clinical Electrophysiology of Vision standards showed slightly reduced amplitudes in both cone and rod responses (Fig. 2E).
Genomic DNA was extracted from peripheral blood of the affected proband. Targeted sequencing of 98 candidate genes (Supplemental Table S1) known to be associated with inherited retinal degenerations, including ABCA4, ELOVL4, MCDR2, and PROM1, was performed. We captured genomic DNAs within these 98 target genes using a SureSelect customized kit (Agilent Technologies, Santa Clara, CA, USA) and prepared libraries using the SureSelect Target Enrichment System protocol. Targeted exome sequencing was performed on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA). Sequencing reads were aligned to the Genome Reference Consortium human genome (build37) using Burrow-Wheeler Aligner (v0.7.5) [5] with an algorithm, which works by seeding alignments with maximal exact matches. Aligned Sequence Alignment Map (SAM)/Binary Alignment Map (BAM) format files were sorted and indexed by SAMTOOLS (v1.2) [6], and duplicated reads were removed by Picard tools (v1.124) (https://broadinstitute.github.io/picard/). Secondary reads alignment was performed following known insertions/deletions (indels) from gold-standard (The Single Nucleotide Polymorphism database [dbSNP] build 138, Mills, 1000 Genome Project phase 1 indels) by Genome Analysis ToolKit (GATK) (v3.3-1) [7]. Finally, base recalibration was performed and annotated by GATK. Single nucleotide variants (SNVs) and indels were calculated by Unified Genotyper in GATK. Each called variant was functionally annotated by ANNOVAR [8]. Based on annotated information, the variants were filtered according to the following criteria: i) located in exonic region, ii) minor allele frequency <0.01 of The Exome Aggregation Consortium (ExAC) [9], iii) damaged in prediction tools (SIFT [10], Polyphen2 [11], GERP++ [12]), iv) clinical significance (ClinVar [13]). Structural variants and copy number alterations were calculated by using a cigar string of reads containing breakpoints and normalized read depth in target regions by in-house tools, respectively. Depth of coverage and statistical analysis were calculated from BAM files by CalculateHsMetrics of Picard Tools.
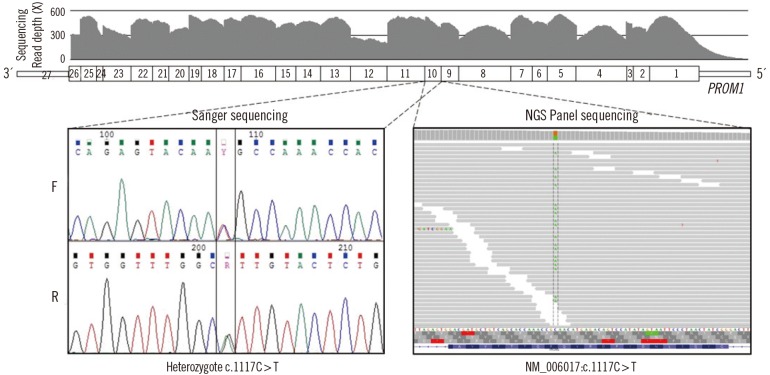
Targeted exome sequencing data were generated with three million sequencing reads, covering 97.8% of targeted genes with a sequencing coverage of ≥100X as well as 99.3% of ≥50X. Targeted regions of PROM1 were fully covered, with a mean depth ranging from 168X to 513X. A heterozygous missense variant in PROM1 (NM_006017.1: c.1117C>T [p.R373C]) was detected in the affected proband with a high read depth. Sanger sequencing using newly-designed forward (5'-GGCTGGATGATGTTTATCAG-3') and reverse primers (5'-GACTCTTGATGACCATATAATG-3') confirmed the heterozygous presence of the variant in the proband (Fig. 3). No pathogenic variants were identified in the other 97 genes. On the basis of the characteristic retinal phenotype and PROM1 p.R373C mutation, the patient was diagnosed as having STGD4.
Classical Stargardt disease (STGD1; OMIM No. 248200), one of the most common hereditary macular dystrophies, is caused by mutations in the ABCA4 gene on chromosome 1p with autosomal recessive transmission [14]. In addition to the autosomal recessive pattern, which is more common, some families with autosomal dominant inheritance have also been described, including STGD3 (OMIM No. 600110), caused by mutations in ELOVL4, and STGD4 (OMIM No. 603786) [1215]. In 1999, STGD4 was mapped to a locus on chromosome 4p [1]. It had an overlap with bull's eye macular dystrophy (MCDR2; OMIM Mo. 608051) [16]. In 2008, mutation analysis of genes within the overlapping disease interval revealed a PROM1 p.R373C mutation [2]. PROM1 is normally localized to the photoreceptor outer segments in the retina. The p.R373C mutation showed defective disk morphogenesis in transgenic mice with this mutation [2]. Ocular phenotypes of the STGD4 families with PROM1 p.R373C mutations may be similar to the phenotypes of STGD1 and STGD3 as well as MCDR2 and cone-rod dystrophy [1234]. Therefore, molecular genetic diagnosis is essential for diagnosing these rare diseases.
Prominin-1 is also known as CD133, which was originally identified as a surface marker of hematopoietic stem cells and endothelial progenitor cells [17]. CD133 is also involved in hippocampal neurogenesis [18]. Although an extraocular phenotype is not remarkable in patients with PROM1 mutation, one study found that ex vivo endothelial function is abnormal in patients with p.R373C PROM1 mutation; One family has exhibited empty sella turcica on brain magnetic resonance imaging and microhematuria [19]. Although the proband in our study had no systemic symptoms or previous medical history, possible systemic involvement associated with PROM1 mutation needs to be examined when the patient shows certain systemic symptoms or signs.
In summary, we report a Korean patient with STGD4 diagnosed by targeted exome sequencing. To the best of our knowledge, this is the first case of STGD4 in Koreans. Targeted exome sequencing seems to be an efficient approach to find genetic causes of macular dystrophy.
 XML Download
XML Download