

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Megalencephalic leukoencephalopathy with subcortical cysts (MLC; MIM 604004) is an autosomal recessive leukodystrophy characterized by infantile-onset macrocephaly and gradual motor deterioration. Infants with MLC are healthy at birth, but macrocephaly develops during the first year of life. Early development is usually normal, but motor disability in the form of spasticity and ataxia manifests slowly [12]. Many individuals with this condition experience epileptic seizures, which are easily controlled with medication. Cognitive function is relatively well preserved. Magnetic resonance imaging (MRI) shows diffuse signal abnormality in cerebral white matter and subcortical cysts in the anterior temporal region.
A causative gene, MLC1, was first identified in 2001 and maps to chromosome 22q13.33 [34]. Approximately 75% of MLC patients have MLC1 mutations, which are inherited in an autosomal recessive manner. Although MLC patients with MLC1 pathogenic variants may show clinical variability, no evidence of genotype-phenotype correlation has been reported [5]. More than 90 different MLC1 mutations have been identified [6789]. There are several reports that suggest founder effects. An ancestral mutation has also been described in Indian Agarwal [10], Jewish [11], Egyptian [12], and Japanese patients [13]. To date, there has been no study on the mutational spectrum of MLC1 in Korean MLC patients.
We performed a comprehensive mutational analysis of MLC1 in five Korean patients with MLC. We found a unique mutational spectrum of MLC1 in these patients by demonstrating a founder mutation. We also presented a case of MLC caused by maternal uniparental disomy (UPD) of chromosome 22.
METHODS
1. Clinical features
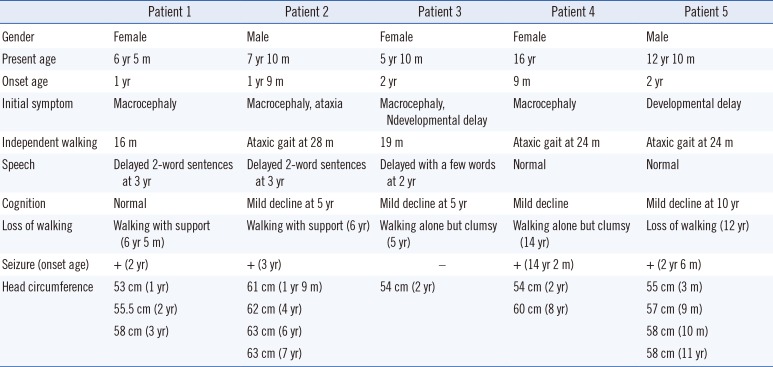
We studied five patients with MLC from unrelated Korean families (Table 1). All five MLC patients were born to nonconsanguineous Korean parents. Initial presentations were macrocephaly and/or motor developmental delay. Macrocephaly was usually detected before 18 months of age (range, 9 months to 2 yr). All patients could walk independently (range, 16–28 months), although most of them showed gait ataxia. Three of the patients exhibited delayed language development and mild declines in cognitive function during the follow-up period, for a median of 8 yr (range, 3–15 yr). Only one patient (Patient 5) was wheelchair bound at the age of 12 yr. Four of the five patients (80%) experienced epileptic seizures, which were easily controlled with antiepileptic medications. All patients had typical MRI findings showing diffuse signal abnormalities in cerebral white matter and the presence of subcortical cysts in the anterior temporal regions (Supplemental Data Fig. S1).
2. Mutational analysis
The study was approved by the institutional review board at Seoul National University Hospital, Korea (IRB No. 1605-133-765). Written informed consent was obtained from all participants. Genomic DNA was obtained from blood samples for each patient and their parents. Direct Sanger sequencing of the entire coding region of the MLC1 (NM_015166.3) was performed. To further evaluate the mutation of Patient 1, we applied multiple ligation-dependent probe amplification (MLPA) (SALSA MLPA P107; MRC-Holland, Amsterdam, The Netherlands) and used a SurePrint G human genome comparative genome hybridization (CGH) plus single nucleotide polymorphism (SNP) 400K microarray (Agilent Technologies, Santa Clara, CA, USA). All procedures were performed according to the manufacturers' protocols. For variant interpretation, we followed the international guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) [14].
3. Genotyping and haplotype reconstruction
We selected 10 SNPs with a minor allele frequency of 0.39–0.50 in this population around the MLC1 region spanning 170 kb as follows: rs36168477, rs5771069, rs137855, rs5771305, rs2010453, rs932330, rs138208, rs731857, rs138244, and rs6010148. Genotyping of 39 unrelated normal controls and five patient families was done by direct sequencing using specific primers for each polymorphic marker. Hardy–Weinberg equilibrium was calculated by using Arlequin (version 3.5.2.2) [15]. Haplotypes were reconstructed by using PHASE (version 2.1) [16].
RESULTS
1. Mutational analysis
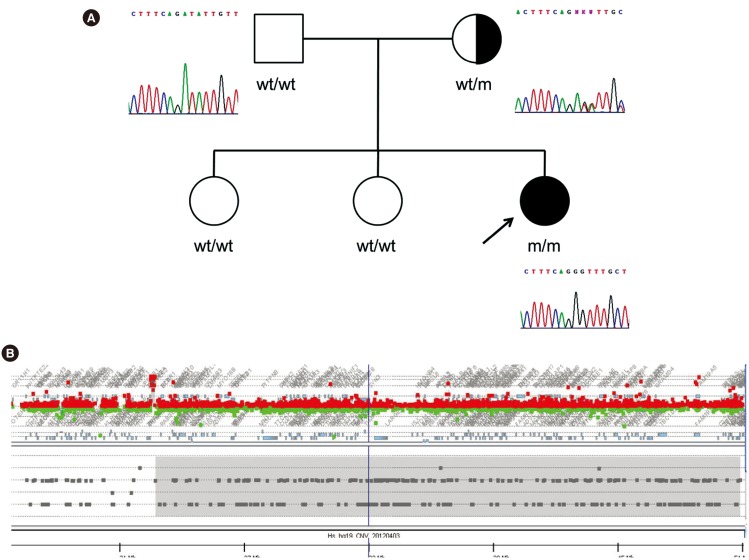
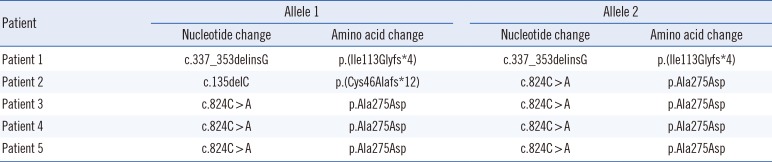
Direct sequencing of the MLC1 in all five patients and available family members revealed three pathogenic variants (Table 2). The p.Ala275Asp variants in exon 10 were observed in seven of 10 alleles (70%). This missense variant was heterozygous in both parents and was inherited in an autosomal recessive manner. Two frameshift variants p.(Cys46Alafs*12) and p.(Ile113Glyfs*4) were also identified. All three variants mentioned were considered pathogenic according to the international guidelines of the ACMG/AMP (Supplemental Data Table S1). Patient 1 was homozygous for the p.(Ile113Glyfs*4) variant. Family studies were performed for this female patient. Only her mother was heterozygous for the p.(Ile113Glyfs*4) variant (Fig. 1A). MLPA did not reveal multiple exon deletions of MLC1. By using the CGH plus SNP microarray analyses, loss-of-heterozygosity of chromosome 22q11.22-q13.33 including MLC1 was predicted. However, no evidence of any large deletion involving the same chromosomal region was identified (Fig. 1B). Therefore, we concluded that a UPD spanning the region of chromosome 22q11.22-q13.33 including the MLC1 was the most plausible mechanism for the homozygous state of the p.(Ile113Glyfs*4) variant in Patient 1.
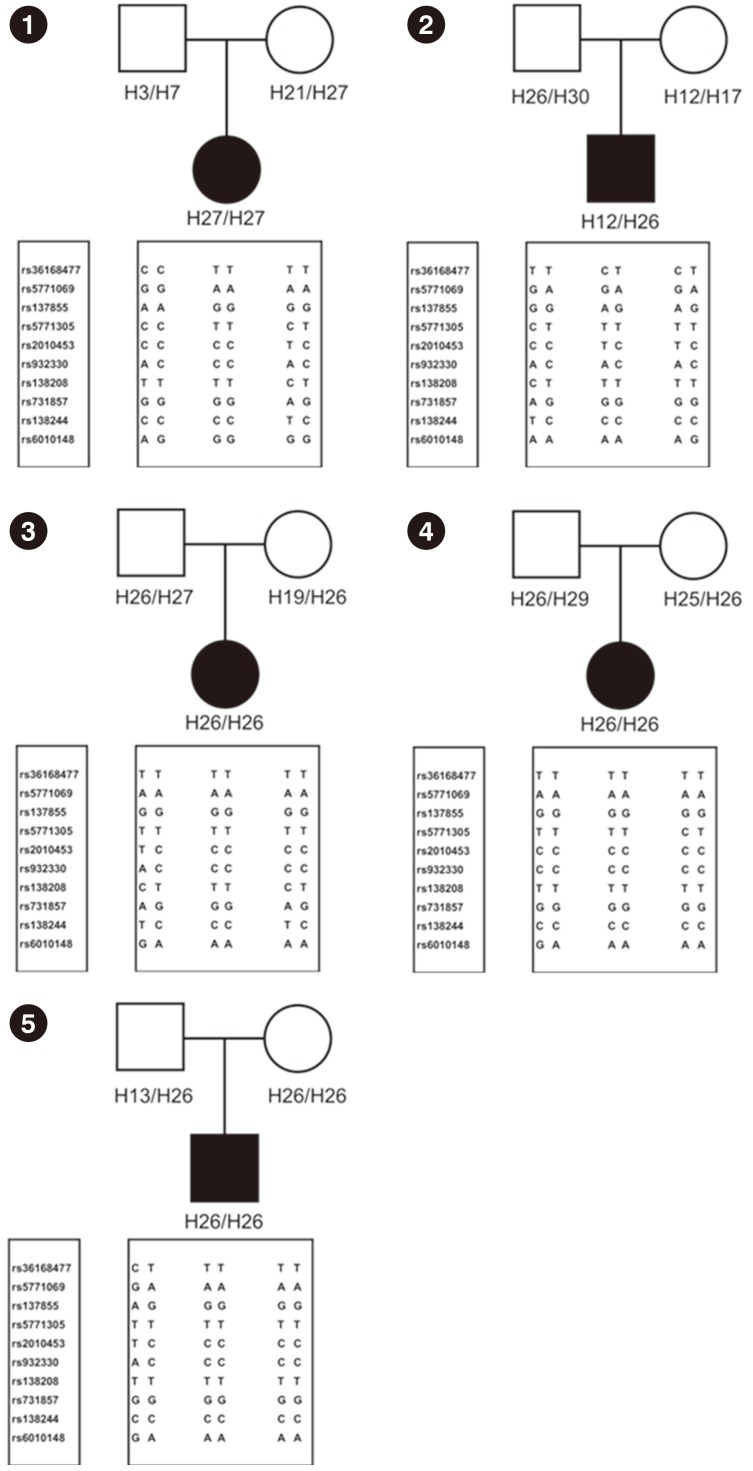
In addition, two chromosomes 22 harboring the MLC1 region were of maternal origin in this patient as shown by haplotype analysis using 10 SNP markers (Fig. 2).
2. Founder effect
Thirty haplotypes were obtained from our genotype data (Supplemental Data Table S2). Haplotype H26 was found in three patients (Patients 3, 4, and 5) with a homozygous p.Ala275Asp variant, and was also found in one patient (Patient 2) along with a heterozygous p.Ala275Asp variant (Fig. 2). Haplotype H26 was segregated within each family, respectively, suggesting that the p.Ala275Asp variant had a common origin.
DISCUSSION
We reported three different MLC1 pathogenic variants from five MLC patients. Seven alleles contained the p.Ala275Asp variant in exon 10. Two frameshift variants p.(Cys46Alafs*12) and p.(Ile113Glyfs*4) were also identified. Using the ACMG/AMP sequence interpretation guidelines [14], we classified all three variants as pathogenic.
The p.Ala275Asp variant was the most prevalent, most common ancestral mutation in the Korean population. Since it was first reported by Montagna et al [17], this variant has been reported in two Japanese MLC patients [1718]. This variant was regarded as the second most common variant in Japanese patients after the p.Ser93Leu variant, which accounts for approximately 80% of Japanese patients with MLC [19]. The p.(Ile113Glyfs*4) variant found in Patient 1 was also reported in one Japanese patient with MLC [12]. These findings suggest a shared genetic background between Japanese and Korean patients with MLC. However, there appears to be clear differences between these two ethnic groups, because the p.Ser93Leu variant was not found in our study.
Patient 1 had a homozygous frameshift variant caused by maternal UPD of chromosome 22. UPD can cause disease by meiotic nondisjunction followed by gamete complementation, trisomy rescue, monosomy duplication, and somatic chromosomal chiasmata [20]. Nondisjunction during meiosis I produces uniparental heterodisomy, in which both homologs of a part of a chromosome from a single parent are passed on to the offspring. By contrast, nondisjunction during meiosis II produces isodisomy, in which two genetically identical copies of a single parental chromosome are inherited [21]. Our results from MLC1 sequencing, MLPA, CGH plus SNP array, and haplotype analysis suggested that UPD of chromosome 22 including the MLC1 region caused MLC in this patient. UPD can cause autosomal recessive disorders, particularly when only one of the parents is a carrier [22]. UPD of chromosome 22 as a disease-causing mechanism has been documented in cases of metachromatic leukodystrophy [23], congenital methemoglobinemia [24], and PLA2G6-associated neurodegeneration [25]. Interestingly, Cao et al [26] recently reported the first case of MLC caused by maternal UPD of chromosome 22. There is no evidence that this could occur more frequently on chromosome 22 than on other chromosomes. However, our case could be an additional example, showing that UPD of chromosome 22 needs to be considered as a possible cause of MLC when conducting MLC1 mutational analysis.
This study is the first to present genetic analyses of Korean patients with MLC. From haplotype analysis, the p.Ala275Asp variant was proven to be an ancestral mutation. We also found that MLC could be caused by maternal UPD, which broadens the documented mutational spectrum of MLC1.
 XML Download
XML Download