

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Peutz-Jeghers syndrome (PJS) is an autosomal-dominant disease characterized by gastrointestinal polyposis and mucocutaneous pigmentation. The gene involved in PJS is mapped to chromosome 19p13.3 and encodes a serine-threonine protein kinase (STK11) known as LKB1 [12]. Here, we report a PJS patient with unique phenotypic features who had heterozygous deletions spanning exons 1–10 of STK11 gene.
A 14-yr-old boy visited the emergency department complaining of acute hematochezia. He had no abdominal pain, history of a change in bowel habits, or any significant loss of weight or appetite. He was the second child of a healthy couple. The boy was delivered at term, by vaginal delivery, after an uncomplicated pregnancy. The first child was an apparently normal boy.
The patient had been followed up at our clinic for routine check-ups since three years of age. He had an atrial septal defect and underwent orchiopexy for cryptorchidism. He had global developmental delay and mental retardation. At about four years of age, black pigment spots appeared on his lips. We suspected PJS and attempted to perform endoscopic investigations several times. However, these failed because he was difficult to sedate, and his vital signs became unstable whenever endoscopy was initiated.
He had a seizure at 11 yr of age with recurrence. There were no characteristics of tuberous sclerosis on brain magnetic resonance imaging (MRI). Electroencephalography showed abnormal, recurrent bilateral frontal high-amplitude sharply contoured delta waves with slowing and intermittent bifrontal sharp and slow complexes. He was started on antiepileptic drugs.
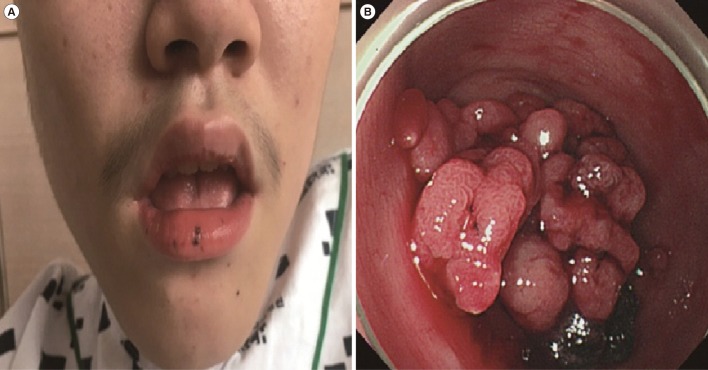
On admission, the abdominal physical examination was unremarkable. A rectal examination revealed black tarry stool, and multiple dark pigmented spots were noted on his lips (Fig. 1A). Initial laboratory tests showed normocytic, normochromic anemia.
Colonoscopy showed a huge grape-shaped pedunculated polyp in the sigmoid colon (Fig. 1B). A few small sessile polyps were found in the terminal ileum, descending colon, and rectum. We suspected that the huge polyp was an active bleeding site. Polypectomy and multiple biopsies were obtained from the polyp. Histologically, the polyp fragments were hyperplastic. No sign of malignancy was observed.
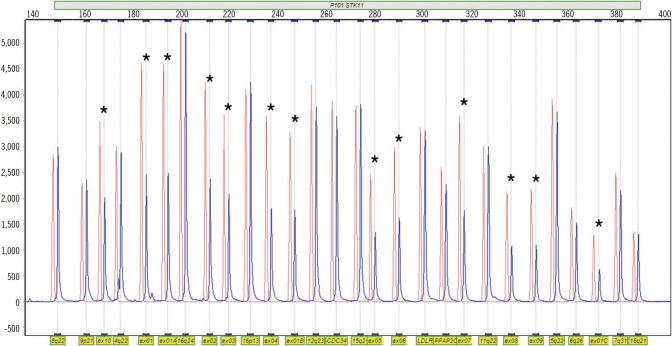
This study was approved by the institutional review board of Soonchunhyang University Bucheon Hospital, Korea. After obtaining informed consent, blood samples were collected from the patient for STK11 analysis. Genomic DNA was extracted from peripheral blood leukocytes. Initially, we performed PCR and direct sequencing using primers targeting the nine coding exons of the STK11 gene. No pathogenic or likely pathogenic variants were detected by this direct sequencing analysis. Next, gene dosage analyses were performed by using a multiplex ligation-dependent probe amplification (MLPA) kit (SALSA MLPA P101-A2 STK11 probemix; MRC-Holland, Amsterdam, Netherlands). Deletion of all 10 exons of STK11 gene was detected (Fig. 2). We performed the same MLPA test in the patient's older brother, with no deletion of STK11 gene detected. His parents refused the gene analysis.
STK11 gene contains nine coding and one noncoding exon, spanning 23 kb, and encoding a 433-amino-acid protein that acts as a tumor suppressor [3]. Approximately 100 different mutations of STK11 in PJS patients have been described [4]. Although large-scale deletions from STK11 have been reported in PJS, including deletion of the entire STK11 gene, these appear to be rare. Whole-gene and exonic deletions and duplications cannot be detected by using conventional screening tests for point mutations and small deletions/insertions. Hence, the prevalence of STK11 mutations in PJS may have been underestimated in the screening studies [5]. The recently developed MLPA technique is a highly sensitive method for detecting copy number changes in genomic DNA sequences and is readily adaptable for high throughput analyses [6]. Therefore, the MLPA assay combined with conventional sequencing methods could increase the detection rate of STK11 mutations.
Some reports have suggested a relationship between the extent of STK11 deletion and various symptoms. Patients with different exonic deletions may possibly have different manifestations because several studies have reported various genotype/phenotype relationships in PJS [7]. However, the STK11 mutation spectrum and genotype-phenotype correlation remains poorly understood [48].
We report a patient whose gene mutation was diagnosed by using MLPA analysis and not by using direct sequencing. This patient's developmental delay, mental retardation, and epilepsy without tuberous sclerosis could be coincidental or possibly related to PJS. This case emphasizes the importance of genetic testing to identify the genotype-phenotype relationship in PJS. The MLPA assay is a valuable method for detecting STK11 mutations in PJS. More studies would be needed to search for large deletions in patients who present with PJS clinically and have atypical clinical features of PJS.
 XML Download
XML Download