

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Choroideremia is a rare X-linked disorder causing progressive degeneration of the retina, retinal pigment epithelium (RPE), and choroid [123]. Affected male patients develop night blindness with progressive peripheral vision loss and central vision loss, usually observed later in their lives [4]. Female carriers may be asymptomatic but can demonstrate patchy chorioretinal atrophy [5]. CHM was identified in the 1990s as a gene responsible for choroideremia. CHM encodes Rab Escort Protein-1 (REP-1), which facilitates posttranslational modification of Rab proteins regulating intracellular trafficking [678]. Various types of mutations in CHM have been identified including small deletions, nonsense mutations, missense mutations, frameshift mutations, splice site defects, and deletion of the entire gene [9]. These mutations cause truncation, loss of functional domain, or absence of REP-1 [10]. In Korea, there has been no report on genetic diagnosis of choroideremia, although a few cases of clinical diagnosis of choroideremia have been reported [1112]. The purpose of this study is to report the first molecular diagnosis of choroideremia in two Korean families, one of which had a novel CHM mutation.
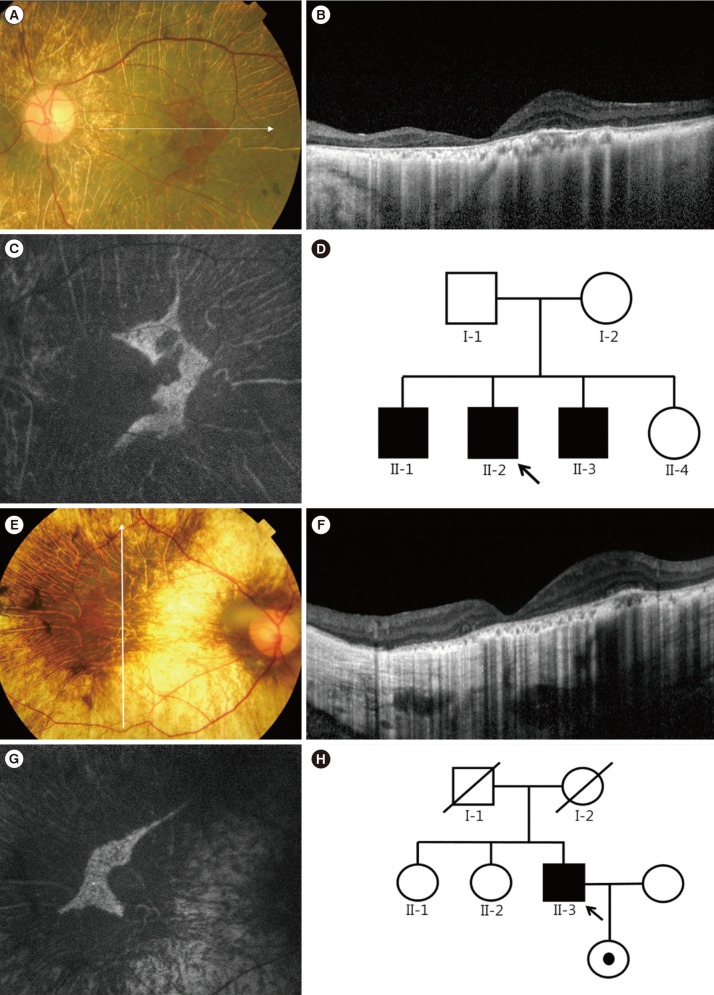
CASE REPORT
In family A, the proband was a 45-yr-old man complaining of night blindness and visual field defect with decreased visual acuity. His uncorrected visual acuity was 20/30 in the right eye and hand motion in the left eye. The proband had been taking immunosuppressant medication subsequent to undergoing kidney transplantation because of chronic glomerulonephritis. In addition, he underwent cataract surgery for posterior subcapsular opacity in both eyes eight years ago. The fundus exam showed bilateral chorioretinal atrophy and areas of RPE disruption with sparing of the central macula (Fig. 1A-D). The residual RPE tissue appeared as a well-demarcated hyperfluorescent area in fundus autofluorescence (FAF) photographs. Standard electroretinograms showed almost extinguished cone and rod responses. An automated visual field test showed a severely constricted visual field in both eyes. Spectral domain optical coherence tomography (SD-OCT) scans showed retinal thinning, choriocapillary atrophy, and abrupt transition to atrophic areas. Increased loss of outer nuclear layer and collapse of outer retina were observed compared to SD-OCT images taken five years ago. The proband's elder brother showed similar symptoms with severe vision loss, which was considered as legal blindness; the ocular phenotype was highly suggestive of choroideremia. In family B, the proband was a 41-yr-old man who was referred to the retina clinic with night blindness and visual field defect in both eyes. His best-corrected visual acuity was 20/40 in the right eye and 20/30 in the left eye. Prior to receiving refractive surgery, his eyes were highly myopic (-10 diopters in both eyes). The findings of the fundus exam, FAF, and SD-OCT scans were analogous to those of obtained for proband A (Fig. 1E-H).
To confirm choroideremia in the probands of family A and family B, genetic analysis of CHM was performed after obtaining informed consent. Genomic DNA was extracted from peripheral blood leukocytes by using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. Fifteen coding exons and their flanking introns were PCR amplified, and the resulting amplicons were sequenced by using an ABI 3730xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) with primers described in Table 1. Targeted sequencing of candidate retinal degeneration genes (including 98 known genes associated with inherited retinal degeneration) was also performed for proband B by using an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) with 101-bp paired-end reads.
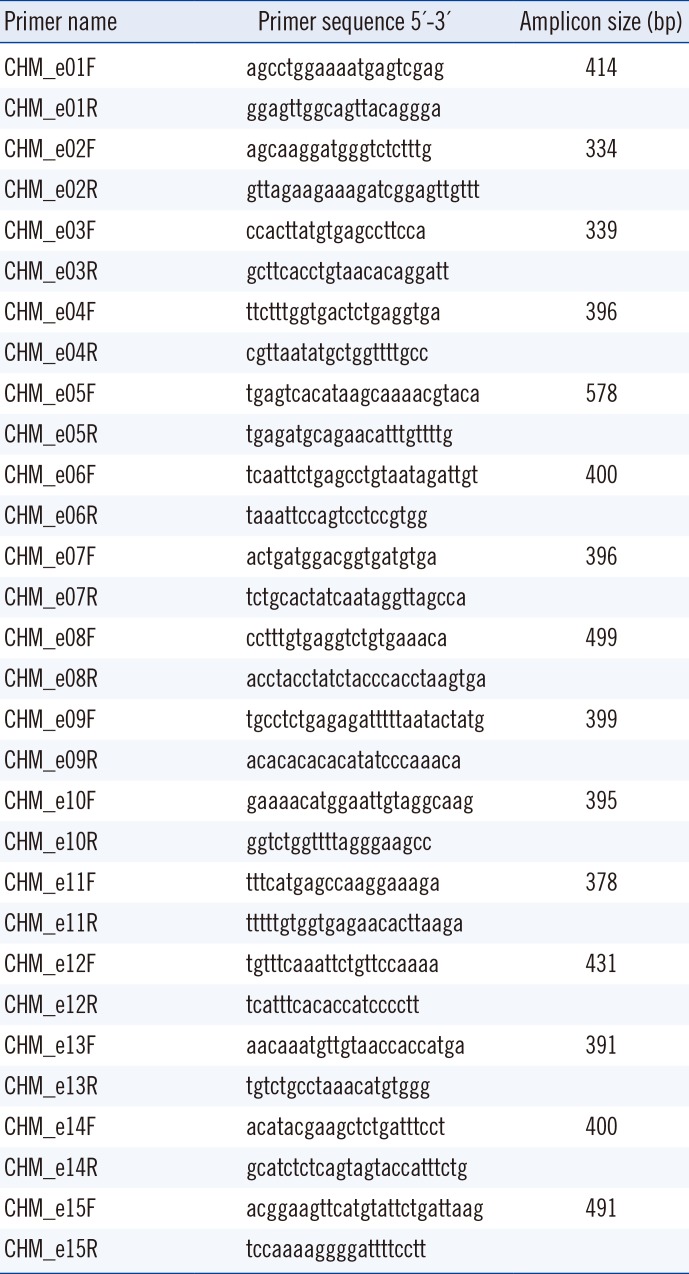
Table 1
Primers used for sequencing analysis of the CHM gene
![]()
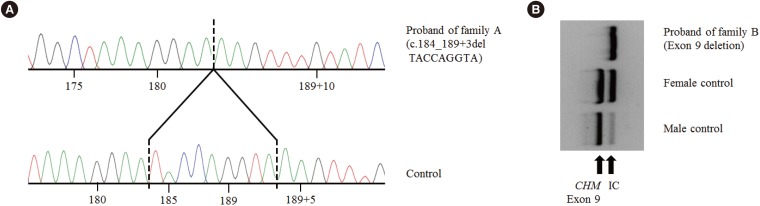
A 9-bp deletion in exon 3 and adjacent intron sequences (c.184_189+3delTACCAGGTA; Fig. 2A) was identified in the proband of family A. The exon 9 PCR product was not detected in the proband of family B, indicating exon 9 deletion (Fig. 2B). Sequencing analysis of other amplified products did not uncover any pathogenic variants; targeted sequencing of 98 candidate retinal degeneration genes in proband B did not reveal any suspicious variations. However, exon 9 of CHM was not captured at all, indicating exon 9 deletion (see Supplemental data Figure S1). No genetic study of proband B's daughter has been performed; however, she is an obligate female carrier of the CHM exon 9 deletion, which is inherited in an X-linked recessive manner.
DISCUSSION
We present two Korean families with choroideremia diagnosed by sequencing CHM. One patient showed a novel small deletion at an exon/intron boundary, and the other revealed a full deletion of exon 9, a known mutation in choroideremia. The novel small deletion variant involves canonical±1 or 2 splice sites, which have been predicted to lead to a null effect and hypothesized to cause disease in choroideremia because the known CHM disease mechanism is loss-of-function. This variant is not present in the data from 622 Korean control exomes or approximately 8,600 East Asian alleles from the Exome Aggregation Consortium (ExAC) [1314]. According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) 2015 guidelines [15], this variant is classified as a likely pathogenic variant based on one very strong piece of evidence (PVS1) and one moderate piece of evidence (PM2). However, an additional family study verifying cosegregation or functional analysis by RNA is needed to confirm the pathogenicity of this variant. Exon 9 deletion has been reported in several choroideremia families and patients [916]. Deletion of exon 9 encompassing nucleotides 1,167 to 1,244 has been predicted to cause a frameshift, leading to a null variant. This deletion is not present in the data from 622 Korean control exomes or approximately 8,600 East Asian alleles from ExAC. Therefore, exon 9 deletion is classified as a pathogenic variant according to the ACMG-AMP 2015 guidelines.
Patients with choroideremia usually show characteristic retinal and choroidal features. However, at times it is difficult to clinically differentiate choroideremia from other inherited retinal degenerations including retinitis pigmentosa and cone-rod dystrophy [1718]. Therefore, a number of researchers have advocated next generation sequencing (NGS)-based approaches and have reported cases of successful diagnosis of choroideremia [17]. However, because choroideremia is often caused by large deletions in CHM, NGS alone may not enable a proper molecular diagnosis for a considerable number of patients with choroideremia. Therefore, when there is a clinical suspicion of choroideremia, a combined molecular genetics approach including direct CHM sequencing, multiplex ligation-dependent probe amplification, and RNA (cDNA) sequencing, as well as NGS-based methods should be considered [19].
In summary, we performed molecular diagnosis of choroideremia in two unrelated Korean families. To the best of our knowledge, this is the first report on the molecular genetic diagnosis of choroideremia in Korean individuals. This study will expand the mutational spectrum of CHM and may help in the development of new treatment modalities such as gene therapy.
 XML Download
XML Download