

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
METHODS
1. Study population
Table 1
Clinical manifestations of Korean patients with GSD Ib with identified SLC37A4 mutations
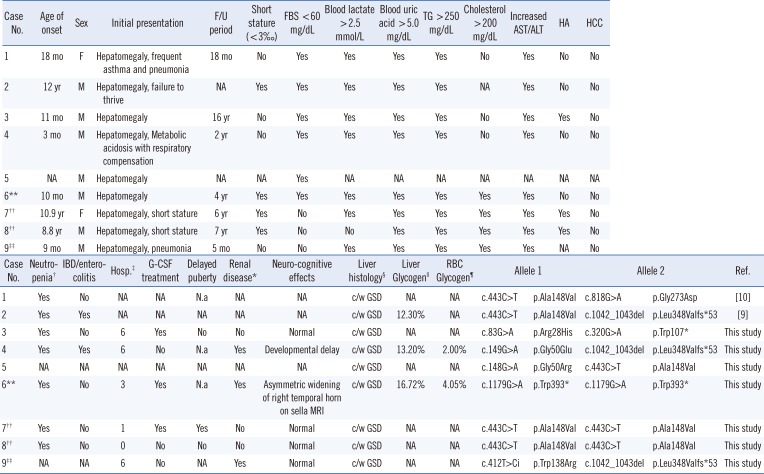
| Case No. | Neutropenia† | IBD/enterocolitis | Hosp.‡ | G-CSF treatment | Delayed puberty | Renal disease* | Neuro-cognitive effects | Liver histology§ | Liver Glycogen|| | RBC Glycogen¶ | Allele 1 | Allele 2 | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Yes | No | NA | NA | N.a | NA | NA | c/w GSD | NA | NA | c.443C>T | p.Ala148Val | c.818G>A | p.Gly273Asp | [10] |
| 2 | Yes | Yes | NA | NA | NA | NA | NA | c/w GSD | 12.30% | NA | c.443C>T | p.Ala148Val | c.1042_1043del | p.Leu348Valfs*53 | [9] |
| 3 | Yes | No | 6 | Yes | No | No | Normal | c/w GSD | NA | NA | c.83G>A | p.Arg28His | c.320G>A | p.Trp107* | This study |
| 4 | Yes | Yes | 6 | No | N.a | Yes | Developmental delay | c/w GSD | 13.20% | 2.00% | c.149G>A | p.Gly50Glu | c.1042_1043del | p.Leu348Valfs*53 | This study |
| 5 | NA | NA | NA | NA | NA | NA | NA | c/w GSD | NA | NA | c.148G>A | p.Gly50Arg | c.443C>T | p.Ala148Val | This study |
| 6** | Yes | No | 3 | Yes | N.a | Yes | Asymmetric widening of right temporal horn on sella MRI | c/w GSD | 16.72% | 4.05% | c.1179G>A | p.Trp393* | c.1179G>A | p.Trp393* | This study |
| 7†† | Yes | No | 1 | Yes | Yes | No | Normal | c/w GSD | NA | NA | c.443C>T | p.Ala148Val | c.443C>T | p.Ala148Val | This study |
| 8†† | Yes | No | 0 | No | No | No | Normal | c/w GSD | NA | NA | c.443C>T | p.Ala148Val | c.443C>T | p.Ala148Val | This study |
| 9‡‡ | NA | NA | 6 | No | NA | Yes | Normal | c/w GSD | NA | NA | c.412T>Ci | p.Trp138Arg | c.1042_1043del | p.Leu348Valfs*53 | This study |
Cases 1, 2, 7, 8, and 9 were confirmed to have variant alleles located in trans in the family DNA analysis.
*Proteinuria, renal stones, nephrocalcinosis, or altered creatinine clearance; †Absolute neutrophil count <1.5×109/L; ‡Infection frequency requiring hospitalization after diagnosis; §Swollen hepatocytes with periodic acid-Schiff-positivity and increased accumulation of glycogen in the cytoplasm on electron microscopy (consistent with glycogen storage disease); ∥Reference range 1–6%/wet liver weight; ¶Reference range <10%/packed red blood cell weight; **SLC37A4 mutations were identified through whole-exome sequencing and confirmed by Sanger sequencing; ††These patients are siblings. Case 7 is the proband of the family, and case 8 is her brother; ‡‡This variant was also identified in his asymptomatic female sibling, who was a heterozygote. She did not carry the other mutation of c.1042_1043del.
Abbreviations: GSD, glycogen storage disease; c/w, compatible with; FBS, fasting blood sugar (glucose); F/U, follow-up; G-CSF, granulocyte-colony stimulating factor; HA, hepatic adenoma; HCC, hepatocellular carcinoma; IBD, inflammatory bowel disease; TG, triglycerides; NA, not available (the information was not submitted); N.a, not applicable because of patient's age or sex; mo, months; hosp, hospitalization; RBC, red blood cell.
![]()
2. SLC37A4 mutation analysis
RESULTS
Table 2
SLC37A4 mutation spectrum in nine Korean GSD type Ib patients
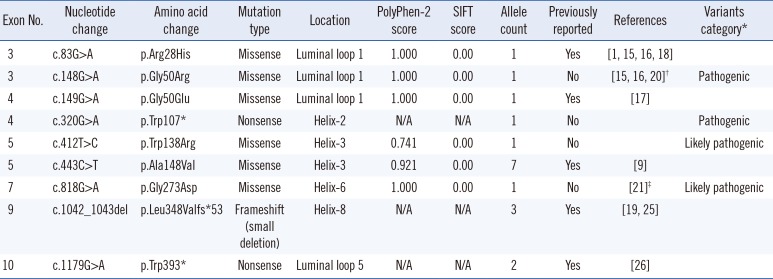
| Exon No. | Nucleotide change | Amino acid change | Mutation type | Location | PolyPhen-2 score | SIFT | Allele count | Previously reported | References | Variants category* |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | c.83G>A | p.Arg28His | Missense | Luminal loop 1 | 1.000 | 0.00 | 1 | Yes | [1, 15, 16, 18] | |
| 3 | c.148G>A | p.Gly50Arg | Missense | Luminal loop 1 | 1.000 | 0.00 | 1 | No | [15, 16, 20]† | Pathogenic |
| 4 | c.149G>A | p.Gly50Glu | Missense | Luminal loop 1 | 1.000 | 0.00 | 1 | Yes | [17] | |
| 4 | c.320G>A | p.Trp107* | Nonsense | Helix2 | N/A | N/A | 1 | No | Pathogenic | |
| 5 | c.412T>C | p.Trp138Arg | Missense | Helix3 | 0.741 | 0.00 | 1 | No | Likely pathogenic | |
| 5 | c.443C>T | p.Ala148Val | Missense | Helix3 | 0.921 | 0.00 | 7 | Yes | [9] | |
| 7 | c.818G>A | p.Gly273Asp | Missense | Helix6 | 1.000 | 0.00 | 1 | No | [21]‡ | Likely pathogenic |
| 9 | c.1042_1043del | p.Leu348Valfs*53 | Frameshift (small deletion) | Helix8 | N/A | N/A | 3 | Yes | [19, 25] | |
| 10 | c.1179G>A | p.Trp393* | Nonsense | Luminal loop 5 | N/A | N/A | 2 | Yes | [26] |
*Previously unreported variants were classified by the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants [14]. †c.148G>A has not been reported previously; however, c.148G>C has been reported as a pathogenic mutation that results in the same amino acid change of p.Gly50Arg. This mutation could be categorized as a pathogenic variant by the ACMG standards and guidelines for the interpretation of sequence variants [14]. ‡c.818G>A has not been reported previously; however, c.817G>A (p.Gly273Ser) has been reported in a GSD patient [21].
Abbreviations: GSD, glycogen storage disease; N/A, not applicable.
![]()
 XML Download
XML Download