

 PDF
PDF ePub
ePub Citation
Citation Print
Print


SCOPE AND PURPOSE
Pharmacogenetics is a rapidly developing field, and the number of pharmacogenetics tests that can be utilized in clinical practice is also increasing. However, the clinical utility of such tests may differ depending on each patient's specific situation and the interpretation of the pharmacogenetic test results. The purpose of these guidelines is to introduce pharmacogenetic tests that can be used in clinical practice and to examine the test application criteria, result interpretation, and reporting methods based on the current literature to enhance the clinical utility of pharmacogenetic tests. We aim to publish the summarized English version here to present its essentials for more readers to understand, although the original version of this article has been published on Laboratory Medicine Online [12]. The pharmacogenetic tests discussed in this guideline are limited to clinical tests covered by the Korea health insurance medical care expenses for patient treatment. Pharmacogenetic tests for the purpose of drug development or research have been excluded.
METHOD OF RECOMMENDATION DEVELOPMENT
These guidelines were developed according to the methodology of the Adaptation Process for Developing Korean Clinical Practice Guidelines Ver. 2.0 [3]. The adapted recommendation was agreed on by the committee and was based on the draft guidelines prepared by integrating selected existing guidelines and literature. The adopted recommendation was peer-reviewed by the development/writing/review committee. It was developed by the Pharmacogenetic Test Clinical Practice Guideline Development Committee at the Korean Society of Laboratory Medicine (KSLM), the Korean Society of Clinical Chemistry, the Korean Association of Quality Assurance for Clinical Laboratories, and the Korean Institute of Genetic Testing Evaluation. This recommendation was developed with the support of KSLM, but the Clinical Practice Guideline Committee worked independently during development/writing/review processes, and KSLM did not influence the development of the recommendation.
To develop the current guideline, the international guidelines related to pharmacogenetic tests for drugs and genes were searched. Among the guidelines published in the past five years, only the guidelines in English were selected, and those written by an individual who did not represent an organization were excluded. In situations where there was no guideline, systematic reviews were the primary targets of the search. Specific searching methods according to the genes are summarized on the clinical pharmacogenetics testing and application: laboratory medicine clinical practice guideline [4]. The comprehensive literature search examined databases such as MEDLINE, PubMed, Cochrane Database of Systematic Reviews, EMBASE, and KoreaMed, where the keywords for the search were the corresponding gene and target drug. Articles with human subjects published in English or Korean were searched, along with additional documentation, such as textbooks, recent publications, theses, and consultations from experts in the corresponding area. After removing duplicate documents, abstracts were reviewed to select documentation, and the actual text was reviewed to select the final target documentation.
These guidelines should be amended in three to five years following the development of new pharmacogenetic testing technologies, changes in the medical environment, and the accumulation of evidence related to pharmacogenetic tests.
GUIDELINES PER GENE
This document contains recommendations for each gene and the rationale. The detailed information refers to LMO 2016 [12].
The recommendations for CYP2C9 & VKORC1, CYP2C19, CYP2D6, and TPMT genotype tests have been prepared based on the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline under the National Institutes of Health's Pharmacogenomics Research Network (NIH PGRN) (CYP2C9 & VKORC1 [5], CYP2C19 [67], CYP2D6 [8910], TPMT [1112]), the Dutch Pharmacogenetics Working Group (DPWG) guideline [13], and the Laboratory Analysis and Application of Pharmacogenetics to Clinical Practice of the National Academy of Clinical Biochemistry (NACB) [14]. Recommendations for UGT1A1 genotype tests were prepared on the basis of NACB's Laboratory Analysis and Application of Pharmacogenetics to Clinical Practice [14], CPIC [15], and the DPWG guideline [13]. Recommendations for NAT2 genotype tests were prepared on the basis of systematic review [16] because there was no representative guideline for NAT2.
The recommendations based on EGFR genotype tests were prepared on the basis of the Molecular Testing Guideline of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology (CAP/IASLC/AMP) [17]. The National Comprehensive Cancer Network (NCCN) guideline [18] was also referenced. The recommendations for HER2 (ERBB2) genotype tests were prepared on the basis of the American Society of Clinical Oncology (ASCO)/CAP Clinical Practice guideline [192021], the NCCN guideline [22], the Spanish Society of Pathology (SEAP), the Spanish Society of Medical Oncology (SEOM) guideline [23], and the United Kingdom (UK) Recommendation [24]. The recommendations for KRAS genotype tests were prepared on the basis of the NCCN guideline [18], the ASCO Provisional Clinical Opinion [25], the European Society for Medical Oncology (ESMO) Clinical Practice guideline [26], the 2010 European Science Foundation-University of Barcelona (ESF-UB) Conference on Pharmacogenetics and Pharmacogenomics [27], the Italy Recommendation [28], and the SEAP and SEOM guidelines [29].
1. CYP2C9 and VKORC1 genes and warfarin
1) Recommendation
For warfarin treatment, it is recommended that CYP2C9 and VKORC1 genotype tests be performed for proper individualized drug dosing.
2) Rationale
Warfarin is the most commonly prescribed oral anticoagulation agent. It has excellent efficacy, but it also has a narrow therapeutic index, and the success of treatment varies among individuals, making it difficult to determine the dose [303132]. There is also a high risk of complications from warfarin treatment; the patient can become over-anticoagulated or under-anticoagulated during this period, leading to a much greater risk of thromboembolism or hemorrhage.
CYP2C9 is a drug-metabolizing enzyme belonging to the cytochrome P450 (CYP) superfamily. This enzyme is expressed in the liver and is the major metabolic enzyme of S-warfarin. Homozygous wild-type CYP2C9*1 shows normal enzyme activity. The most common variants with reduced enzyme activity in the Western population are CYP2C9*2 (rs1799853) and CYP2C9*3 (rs1057910) [33]; in the Asian population, including Koreans, there have been no reports of CYP2C9*2 (rs1799853) [3334]. CYP2C9*2 and CYP2C9*3 have been reported to reduce S-warfarin metabolism by about 30–40% and 80–90%, respectively, in in vitro and in vivo studies [33]. Patients with CYP2C9*2 or CYP2C9*3 alleles are at a greater risk of hemorrhage during warfarin treatment [303536] than those with homozygous wild-type CYP2C9*1 alleles, and therefore should receive a lower dose. Patients with CYP2C9*2 or CYP2C9*3 alleles also require more time until the prothrombin time international normalized ratio (PT INR) is stabilized [3537].
The VKORC1 gene encodes the vitamin K epoxide reductase, which is the target enzyme of warfarin [3839]. Vitamin K epoxide reductase is involved in reducing vitamin K epoxide to vitamin K, which is the rate-limiting step of the vitamin K circuit [40]. The typical non-coding variant c.-1639G>A (VKORC1 G3673A, rs9923231) changes a transcription factor binding site in the VKORC1 gene and thus reduces gene transcription [3241], which is closely related to the low warfarin dose requirement [323441424344].
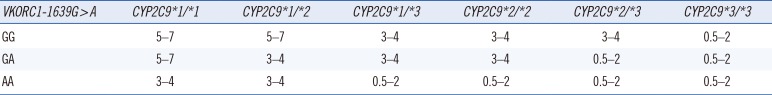
In Asian populations, including Koreans, the CYP2C9*3 variant should be the first CYP2C9 variant tested. For the VKORC1 gene, VKORC1 -1639G>A should be tested directly or the c.174-136C>T (VKORC1 1173C>T, rs9934438) variant should be tested because it exhibits complete linkage disequilibrium with the VKORC1 -1639G>A variant. Allele frequencies of CYP2C9 and VKORC1 in the Korean population are 0% for CYP2C9*2, 4–5% for CYP2C9*3, and 87–94% for VKORC1 1173C>T.
The average maintenance dose based on CYP2C9 and VKORC1 genotype as recommended by the U.S. Food and Drug Administration (FDA)-approved warfarin (Coumadin) labeling is summarized in Table 1. Within each dose range, the patients' age, body surface area, interacting drugs, and other major factors should be considered when determining drug dosage. Additionally, when administering warfarin, application of a genotype-based dosing algorithm that determines the warfarin maintenance dose based on CYP2C9 and VKORC1 genotypes is recommended [45]. The International Warfarin Pharmacogenetics Consortium (IWPC) algorithm [44] and Gage [43] algorithm are commonly used. These algorithms include genotype information and clinical information so that the warfarin maintenance dose required in a stable state can be more accurately predicted [4344]. Such genotype-based dosing algorithms have a greater predictive power for the proper maintenance dose than the fixed dose method or the traditional clinical algorithm.
Most genotype-based dosing algorithms target PT INR 2–3, so it is difficult to apply these algorithms when the target INR lies outside this range [46]. Even if drug is administered according to the genotype-based dosing algorithm, PT INR must be monitored [5]. For children, there are almost no data regarding the related algorithms or the influence of CYP2C9 and VKORC1 genotype on the warfarin dose [47]. There is no warfarin dosing recommendation for children. The benefit of genotype testing is low for patients who have stably taken warfarin over a long period, for patients having difficulty achieving a stable dose due to poor compliance, or for patients who are on a diet including vitamin K. Genotype testing to determine the correct dose is most effective before treatment begins or at the initial stages of treatment [48].
2. CYP2C19 gene and clopidogrel
1) Recommendation
The use of CYP2C19 genotype tests is recommended in cases of anti-platelet agent treatment in patients with acute coronary artery syndrome who have received or are planning to receive percutaneous coronary intervention.
2) Rationale
Clopidogrel is converted to its active form by various CYP enzymes to suppress platelet aggregation, leading to an antiplatelet effect. In particular, activation of the CYP2C19 enzyme determines the rate of conversion from clopidogrel into its active form. Thus, the incidence of stent thrombosis and major complications of acute coronary artery syndrome differs among patients [7]. When clopidogrel is used for treatment, lower CYP2C19 activation leads to fewer circulating active metabolites, thus reducing its suppressive effect on platelet aggregation. Therefore, the use of CYP2C19 genotype testing is recommended to predict a patient's response to clopidogrel or to consider alternative drugs.
Homozygous wild-type CYP2C19*1 has normal enzyme activity. Individuals with heterozygous and homozygous CYP2C19*2 genotypes have 1.55 times and 1.76 times greater risk of suffering from major cardiovascular diseases, respectively, than patients with homozygous wild-type CYP2C19*1 [49]. Additionally, stent thrombosis likelihood is increased by 2.67 times in individuals with the heterozygous variant and by 3.97 times in those with the homozygous variant [49]. In conditions, such as atrial fibrillation, cerebral infarction, or stable angina, an association between reduced activities from CYP2C19 gene variants with the risk of cardiovascular side effects has not been established. It has been reported in the West that the frequency of the CYP2C19*3 variant is low, but this variant does reduce the response to clopidogrel [50]. In studies on Chinese and Korean populations, the CYP2C19*3 variant was reported to reduce the reactivity to levels similar to those of the CYP2C19*2 variant [515253545556].
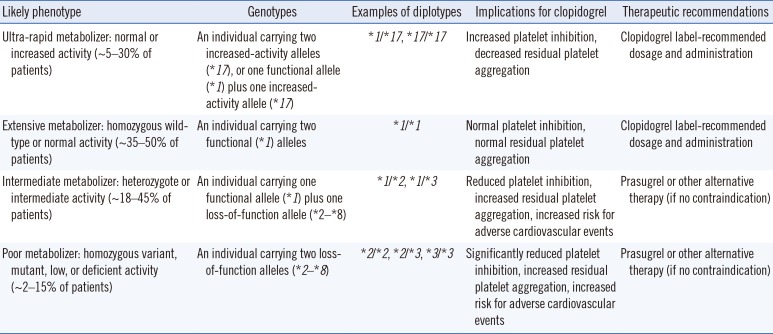
Variants in CYP2C19 are found across the whole gene, but since typical variants causing enzyme dysfunction are known, it is reasonable to test these alleles first. The CYP2C19*2 and CYP2C19*3 alleles are the most frequent, but most alleles of the CYP2C19 gene reduce the metabolic rate of clopidogrel. On the other hand, the CYP2C19*17 allele increases the metabolic rate of clopidogrel. The frequency of CYP2C19*3 in the Korean population is relatively high, at 7–10%, but this variant is rarely detected in the West. In contrast, the frequency of CYP2C19*17 in Koreans, which is relatively common in the West, is very low, at about 1%. The phenotypes resulting from the CYP2C19 genotypes are classified into four categories (Table 2): extensive metabolizer (EM), poor metabolizer (PM), intermediate metabolizer (IM), and ultra-rapid metabolizer (UM). The phenotype is categorized as EM for homozygous wild-type CYP2C19*1, IM for CYP2C19*1 and CYP2C19*2/*3 heterozygous variants, PM for two dysfunctional alleles (CYP2C19*2, CYP2C19*3, etc.), and UM for the homozygous CYP2C19*17 variant or heterozygous CYP2C19*1 and CYP2C19*17 variants.
For patients classified as PM or IM by CYP2C19 genotype tests, the administration of recently developed antiplatelet agents such as prasugrel or ticagrelor can be considered instead of clopidogrel (Table 2). In IM cases, it has been reported that during clopidogrel treatment, the remaining platelet activity is increased and the risk of serious cardiovascular side effects is increased; however, because interindividual variation is significant, it is best to consider other clinical factors. There is not yet enough evidence on the effects of increased clopidogrel dose in patients expected to show reduced clopidogrel metabolism based on CYP2C19 genotype analysis [7]. When the use of proton pump inhibitors is combined with clopidogrel, CYP2C19 enzyme activity is suppressed, and the effect of clopidogrel can be influenced by reducing the generation of the active form of clopidogrel [756].
3. CYP2D6 gene and tricyclic antidepressants, codeine, tamoxifen, and atomoxetine
1) Recommendation
When administering tricyclic antidepressants, codeine, tamoxifen, or atomoxetine, the use of CYP2D6 genotype test is recommended.
2) Rationale
The functionality of CYP2D6 in drug metabolism can be determined on the basis of patient's CYP2D6 genotype. The side effects and safety of tricyclic antidepressants, codeine, tamoxifen, and atomoxetine vary according to differences in the metabolism capacity of CYP2D6 based on the CYP2D6 genotype of the individual patient [89101457]. The drug removal rate or conversion rate to active metabolites also differs depending on CYP2D6 activity, thus, the risks of treatment failure or side effects may increase in different CYP2D6 variants.
Tricyclic antidepressants are compounds that inhibit the reabsorption of serotonin and norepinephrine; they are metabolized into hydroxyl metabolites with low activity by CYP2D6 [10]. Tricyclic antidepressant metabolism is delayed in CYP2D6 PM so that the blood tricyclic antidepressant concentration increases, leading to the increased possibility of side effects, such as anticholinergic actions, central nervous system disorders, heart function disorders, and other conditions. In UM, decreased blood tricyclic antidepressant concentration leads to the possibility of treatment failure [10].
Codeine is a narcotic analgesic. About 80% of the administered codeine is converted into inactive metabolites, and about 5–15% is metabolized by CYP2D6 into the active metabolite morphine. Therefore, CYP2D6 metabolism is important for the analgesic effect of codeine [89]. Codeine conversion rate to morphine is significantly decreased in CYP2D6 PM, so that the analgesic effect may be insufficient, whereas the rate of conversion to morphine is increased in CYP2D6 UM, possibly increasing drug side effects such as nausea, vertigo, drowsiness, sedation, shortness of breath, constipation, pruritus, difficulty in breathing, circulatory disorders, apnea, shock, or heart attack [89].
Tamoxifen is a selective estrogen receptor modulator used for treating and preventing hormone-dependent breast cancer. Tamoxifen is a prodrug, and CYP2D6 is involved in its metabolism into endoxifen, which is the rate-limiting step. Endoxifen is an active metabolite with 30–100 times higher antiestrogen activity than tamoxifen. It has been suggested that variations in CYP2D6 are involved in the individual differences in drug metabolism and response among patients, but there are inconsistent results across studies [145758]. The blood endoxifen concentration decreases owing to the reduced conversion of tamoxifen to endoxifen in CYP2D6 PM, accordingly, the risk of breast cancer recurrence is increased. In UM patients, the concentration of the active metabolite increases, so the possibility of side effects is increased [1314]. There are large interindividual variations in endoxifen concentration, and this is thought to be partially due to CYP2D6 gene variation [145758].
Atomoxetine is used to treat attention deficit hyperactivity disorder (ADHD) in children, teenagers, and young adults. Atomoxetine is metabolized by CYP2D6 into hydroxyatomoxetine with low activity. The effect of atomoxetine may be reduced in CYP2D6 UM patients. In PM, the half-life is significantly increased, so atomoxetine concentration is about five times higher than that of EM [14]. Therefore, the possibility of adverse drug reactions such as headache, insomnia, xerostomia, abdominal pain, nausea, reduced appetite, or coughing increases. When taken with CYP2D6 inhibitors, such as paroxetine or fluoxetine, the atomoxetine metabolism is suppressed, so that adverse effects similar to those associated with CYP2D6 PM may occur [14].
To determine the CYP2D6 genotype, a test for large gene deletions/duplications, such as long PCR or multiplex ligation-dependent probe amplification (MLPA), should be conducted alongside a test for single-nucleotide variants. When testing in Koreans, a method that can detect CYP2D6*10, gene deletions (CYP2D6*5) and amplification (xN) should be included.
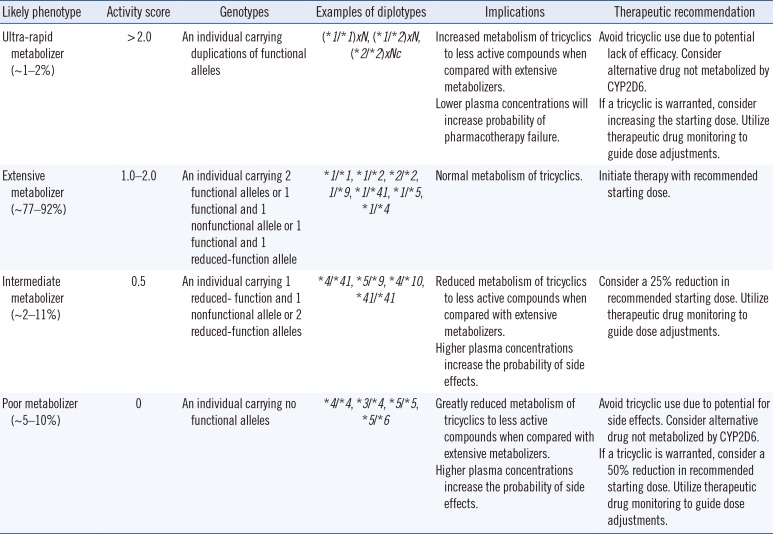
Based on the activity score calculated from the combinations of CYP2D6 alleles, the CYP2D6 phenotype can be determined [5960]. The recommendations for tricyclic antidepressant treatment based on CYP2D6 phenotype are shown in Table 3.
The codeine treatment recommendations based on CYP2D6 phenotype are as follows: in UM, owing to the high possibility of adverse drug effects, the use of codeine should be avoided, and the use of an alternative analgesic agent should be considered. In IM, treatment is initiated at the usual dose, and the use of an alternative analgesic agent should be considered if pain is not mitigated. In the case of PM, the drug effect could be reduced, so the use of an alternative analgesic agent should be considered [8].
For tamoxifen, established administration recommendations are not available, but the DPWG guideline recommends an aromatase inhibitor for post-menopausal breast cancer patients [13], on the basis of clinical studies [6162].
In case of atomoxetine in EM, the recommended atomoxetine dose is 1.2 mg/kg/day for a body weight of 70 kg or below and 80 mg/kg/day for a body weight of 70 kg or above. In PM, the recommendation is 0.5 mg/kg/day for a body weight of 70 kg or below and 40 mg/day for a body weight of 70 kg or above [14].
4. NAT2 gene and isoniazid
1) Recommendation
When administering the anti-tuberculosis drug isoniazid, the use of NAT2 genotype tests is recommended.
2) Rationale
Isoniazid is one of the first-line treatment regimens for tuberculosis. It is metabolized primarily in the liver by arylamine N-acetyltransferase 2 (NAT2). Hepatitis is a common adverse effect of isoniazid administration. The risk of drug-induced hepatotoxicity varies depending on acetylation status determined by NAT2 gene alleles [636465].
The wild-type NAT2 allele is NAT2*4 [66]. The Korean population shows a lower frequency of the NAT2*5 allele, which is common in other races, but it has a relatively high frequency of the NAT2*6 (c.590G>A) and NAT2*7 (c.857G>A) alleles, which show reduced NAT2 enzyme activity. The phenotypes are divided into three types according to the number of alleles, and the enzyme activity of each genotype is shown in Table 4. The cases with two rapid alleles (such as NAT2*4), one rapid allele and one slow allele (such as NAT2*6 or NAT2*7), and two slow alleles are classified into the rapid acetylators, intermediate acetylators, and slow acetylators, respectively. Also, cases with at least one or more wild-type allele (NAT2*4) are classified into the rapid acetylator group, and all other cases or the cases with no NAT2*4 allele are classified into the slow acetylator group in other publications [1667]. Phenotype frequencies of NAT2 gene variations in the Korean population are 39–45% for rapid acetylators, 44–50% for intermediate acetylators, and 8–11% for slow acetylator [6869].
NAT2 activity is determined by the number of active alleles. Decreased acetylation is associated with a reduced isoniazid clearance rate, thus increasing exposure to the drug and its related metabolites. The risk of drug-induced hepatotoxicity is higher in the slow acetylator group, in which the activity of both NAT2 alleles is reduced, than it is in the rapid acetylator group, which has normal allele activity [167071]. Meta-analysis has shown that, although the odds ratio for the drug-induced hepatotoxicity of the NAT2 slow acetylator group has been reported to be 3.32 (95% confidence interval 2.43–4.53) in East Asians, no correlation has been found in Caucasians [16].
There is currently no agreement on appropriate isoniazid dosing based on NAT2 genotype or phenotype because of few clinical studies; however, a recommendation for dosing based on the genotype may be used if data are accumulated in the future. In a recent randomized control study in Japan and Korea, NAT2 genotype and phenotype-based isoniazid dosing for an anti-tuberculosis therapy was suggested [7072]. In the slow acetylation group, there was no loss in treatment efficacy when a reduced dose was administered, but drug-induced hepatotoxicity decreased. In the rapid acetylation group, the initial treatment failure rate decreased without increased hepatotoxicity when increasing the dose. So far, however, there has been no established recommendation, and future studies are necessary. During anti-tuberculosis treatment, measuring blood drug concentration may be helpful when the effects of treatment are inadequate as a result of drug absorption, administration compliance, and combined medication influence [7374].
5. UGT1A1 gene and irinotecan
1) Recommendation
For high-dose irinotecan therapy (>250 mg/m2), the use of UGT1A1 genotype testing is recommended before treatment to help determine the proper initial dose and prevent drug-related adverse reactions [14]. According to the recent review by French joint working group, pretreatment UGT1A1 genotyping is recommended for all patients scheduled to receive an irinotecan dose ≥180 mg/m2 [75].
2) Rationale
Irinotecan is used for treating metastatic colon cancer and other solid cancers. It is glucuronated by the uridine diphosphate glucuronosyl-transferase (UGT) 1A1 enzyme in liver cells and is excreted into bile in its inactive state. When UGT1A1 enzyme activity is decreased, the active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38) may cause irinotecan-related adverse effects due to decreased metabolism and excretion.
Owing to the variation in drug metabolism when administering irinotecan based on the UGT1A1 genotype, serious side effects such as severe neutropenia or diarrhea may occur. It has been reported that the relative risk of neutropenia is 2.0–7.2 times higher in people homozygous for the UGT1A1*28 allele than in those with the homozygous wild-type genotype, and this risk increases with a higher dose of irinotecan [76]. The UGT1A1*6 allele, which is relatively common in Asians, is also related to irinotecan-induced toxicity [7778]. Therefore, in Asian patients, it is desirable to check for both UGT1A1*28 and UGT1A1*6 when conducting UGT1A1 genotype tests. The allele frequencies of UGT1A1 in the Korean population are 18.6–22.2% for UGT1A1*6 and 9.5–12.7% for UGT1A1*28 [798081]. Genotype tests should be used in addition to treatment policy determination and cannot replace the clinician's decision and clinical experience. Additionally, other important factors such as liver function, kidney function, age, and concurrently used medicine, should be considered.
The enzyme activity according to each UGT1A1 genotype and phenotype is shown in Table 5. When administering a high dose of irinotecan (250 mg/m2 or greater) to patients who are homozygous for the UGT1A1*28 variant allele, administration of a dose that is 1 degree lower (e.g. 125 mg/m2) [14] or a dose that is decreased by 30% is recommended [13]. For patients with the UGT1A1*28 heterozygous variant, reducing the dose may cause under-treatment, so it is not recommended [13]. UGT1A1*6 also reduces the activity of the UGT1A1 enzyme, but there has not been an established recommendation on drug dose control in patients with UGT1A1*6 [82].
6. TPMT gene and thiopurine family drugs (azathioprine, mercaptopurine, and thioguanine)
1) Recommendation
TPMT genotyping before administering thiopurines (azathioprine, mercaptopurine, and thioguanine) is recommended to determine the proper initial dosing.
2) Rationale
Azathioprine is used mainly to treat immune diseases, including inflammatory bowel disease, mercaptopurine is used to treat immune diseases and lymphoid malignancies, and thioguanine is used to treat myelogenous leukemia. Azathioprine is metabolized into mercaptopurine, most of which is converted into the inactive form methyl-mercaptopurine (methylMP) by thiopurine S-methyltransferase (TPMT). A small amount of mercaptopurine goes through multiple stages of metabolism, being converted into methyl-thioinosine monophosphate (methylTIMP), which results in immunosuppression and hepatotoxicity, and the major active form of thioguanine nucleotide (TGN) [118384].
The lower the TPMT activity, the more the blood TGN level increases when thiopurines are used. In homozygous variants that carry two nonfunctional TPMT alleles, administering a conventional dose of azathioprine and mercaptopurine maintains a high TGN concentration and thus results in severe life-threatening myelosuppression (myelosuppression risk 100%). In cases of heterozygotes with a single nonfunctional TPMT allele, there is a moderate to severe myelosuppression risk (30–60%). In homozygous wild-type patients with two functional alleles, the risk of myelosuppression is relatively low [1285]. When using thiopurines in patients with low TPMT activity, late-developing adverse effects such as secondary cancers may occur even if severe myelosuppression does not occur. There have been no reports associating the adverse effects of reduced TMPT activity with drugs other than thiopurines, and pancreatitis and hepatotoxicity have not been found to be related to low TMPT activity [12].
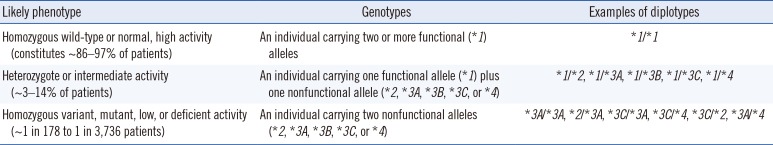
Although 30 or more TPMT alleles are known, only a few alleles that cause reduced enzyme activity are prevalent. Thus, common alleles can be preferentially selected to perform TPMT genotyping [86]. The TPMT*3 has the highest prevalence (0.8–2.5%) in the Korean population, but the presence of *6, *16, *32, and *38 has been reported [87]. The TPMT phenotypes based on each genotype are described in Table 6.
When dosing is appropriately controlled on the basis of TPMT genotype and phenotype, the anticancer and immunosuppressive effects of thiopurines can be maintained while reducing the adverse effects. Toxicity can also be reduced without increased risk of recurrence in patients with acute lymphoblastic leukemia. Although the number of patients with TPMT variants is small, there are many cases where thiopurines are administered alongside other drugs, inducing myelosuppression. These cases may result in severe adverse effects such as fatality or myelosuppression even after a short conventional dose of thiopurines in patients with homozygous variants. Therefore, TPMT genotyping before initiating thiopurine treatment is recommended to determine the initial dose.
In cases where a conventional dose is administered in TPMT heterozygous variants, only 30–60% of patients show myelosuppression, and administering a reduced dose to all patients leads to the undertreatment of a portion of patients who would not have shown adverse effects. Therefore, to minimize undertreatment until the steady state is reached, the patient's state, concomitant medications, adverse effects, and disease progression should be monitored to adjust the dose. Therapeutic drug monitoring (TDM) can be performed by measuring the thiopurine metabolites of TGN and the methylMP in peripheral blood erythrocytes. This can help to evaluate treatment compliance and the influence of concomitant medications, as well as to assist in judging whether thiopurine administration decreases leukocytes [1288].
7. EGFR gene and non-small-cell lung cancer
1) Recommendation
To select non-small-cell lung cancer patients who are candidates for tyrosine kinase inhibitor (TKI) treatment—a treatment agent for targeting the epidermal growth factor receptor (EGFR)—the use of EGFR gene testing is recommended.
2) Rationale
The European Medicines Agency stipulates that EGFR gene testing must be performed when using TKIs, especially gefitinib. According to Korean Association for Lung Cancer (KASLC) clinical guidelines, in non-small-cell lung cancer cases where there is an EGFR gene mutation, gefitinib or erlotinib is prescribed as the primary anticancer treatment. In cases where there are no mutations, platinum-based chemotherapy is recommended. The usage precautions for gefitinib (Iressa) outlined by the Korea Food and Drug Administration state that it is important to select a properly verified and established method to avoid false negative or false positive results when evaluating a patient's EGFR mutation state.
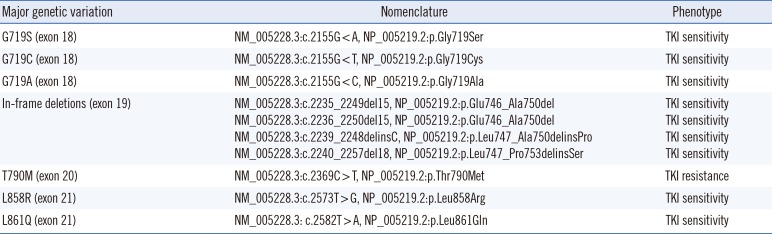
A non-small-cell lung cancer patient with an active mutation in exons 18-21 of the EGFR gene will respond to TKIs, and their average life expectancy is extended. An in-frame deletion in exon 19 (amino acids 729–761) and L858R located in exon 21 are the most common mutations that result in drug sensitivity, and these are responsible for approximately 90% of cases [1718]. A significant number of patients react to initial TKI treatment, however, they acquire drug tolerance during treatment. T790M, located in exon 20, is associated with acquired resistance. The T790M mutation is rarely detected in patients before treatment. In rare cases, it can be discovered as a germline mutation in cases of familial lung cancer [1718].
The EGFR mutation is present in Asians at a frequency of approximately 30–40% and in Caucasians at a frequency of approximately 10–15%. EGFR mutations also are frequent in non-smokers and females. However, clinical information (e.g., smoking, gender, or race) is not sufficiently sensitive to select patients for TKI treatment or EGFR testing, and it is recommended that such testing be performed on all target patients [1718].
EGFR genetic testing can be performed by using paraffin-embedded, fresh, frozen, or alcohol-fixed tissue or body fluid specimens. A pharmacogenetic method that can detect mutations in specimens with at least 50% tumor cells must be used [17]. The use of a testing method that can detect mutations in specimens with as little as 10% tumor cells is strongly recommended. To detect acquired T790M mutations, the testing method should be able to detect the mutation in as few as 5% of cells [17]. Such a method should be able to detect all gene mutations that have been reported to be showing a frequency of at least 1% of EGFR-mutated lung cancers. In the case of testing with the purpose of selecting TKI targets, immunohistochemistry (IHC) is not recommended.
One should ascertain if the detected mutation is an active mutation or a resistance mutation based on the TKI phenotype for each EGFR mutation organized in Table 7.
8. HER2 gene and breast cancer
1) Recommendation
To determine whether to use HER2 targeted therapy in breast cancer patients, in situ hybridization (ISH) is recommended to determine HER2 gene amplification or IHC is recommended to check HER2 protein overexpression.
2) Rationale
HER2 overexpression is observed in 15–20% of breast cancer cases, and cases positive for HER2 overexpression respond to HER2-targeted treatment [20]. Thus, the presence of HER2 overexpression can determine the therapeutic response to HER2-targeted treatment. Therefore, in patients with invasive breast cancer, it is recommended that HER2 gene amplification or HER2 overexpression testing be performed before anti-HER2 therapy, and that only those patients who exhibit HER2 overexpression be administered anti-HER2 therapy.
HER2 is a gene that encodes proteins belonging to the EGFR family of tyrosine kinase receptors, and this gene is also called HER2, NEU, NGL, TKRI, Her-2, c-erb B2, and Her-2/new. HER2 gene amplification is the major mechanism that leads to HER2 overexpression; there is a rise in the concentration of 185 kDa glycoproteins with tyrosine kinase activity in tumors with HER2 gene amplification. HER2 overexpression is related to the clinical outcome of breast cancer patients, and it is a powerful predictor for determining the response to the anti-HER2 therapy. In case of HER2 overexpression, anti-HER2 therapies, such as trastuzumab (Herceptin), lapatinib, pertuzumab, or trastuzumab emtansine (KADCYLA), can be used. Therefore, it is recommended that HER2 testing be performed for all patients with invasive (early stage or recurrent) breast cancer and that anti-HER2 therapy be applied only to those cases with positive test results.
HER2 testing uses cancer tissue of the primary or recurring cancer or metastatic area, obtained through an operation or needle biopsy. The time between tissue acquisition and fixation should be minimized (within one hour), and the specimen should be fixed in neutral buffered formalin for 6-72 hr. Tissue sections should not be used for HER2 testing if cut more than six weeks earlier [20].
HER2 testing uses both IHC and ISH. IHC is used to examine HER2 expression, and fluorescent in situ hybridization (FISH) or silver in situ hybridization (SISH) is used to examine gene expression. If pre-analytical or analytical requirements are not satisfied or quality assurance fails, the test should be considered inappropriate for HER2 assessment.
HER2 testing results are either positive, equivocal, negative, or indeterminate. HER2-positive is defined as cases where protein overexpression was determined by IHC by observing expression in at least 10% of contiguous and homogenous tumor cells within the tumor area or in cases where gene amplification was observed by ISH (HER2 copy number or HER2/CEP17 ratio based on a cell count of 20 or more within the area) has been confirmed.
Patients with positive results from HER2 testing can undergo HER2-targeted treatment. When the result of the primary test is ambiguous, the same specimen should be checked through an alternative testing method (IHC or ISH) or testing should be performed on an alternative specimen. If there is a histopathological discordance, additional HER2 testing should be considered, and the decision-making process and results should be reported.
9. KRAS gene and metastatic colorectal cancer
1) Recommendation
In metastatic colorectal cancer patients, the use of the KRAS gene test is recommended to predict the therapeutic effects of anti-EGFR-targeted drugs (cetuximab and panitumumab).
2) Rationale
In about 40% of colorectal cancer cases, mutations are found in codons 12 and 13 in exon 2 of the KRAS gene. If these mutations are present, there will likely be no response to EGFR-targeted treatment. Therefore, it is strongly recommended that the KRAS gene test is performed on the cancer tissue (primary cancer tissue or metastatic cancer tissue) of all stage 4 metastatic colorectal cancer patients when beginning treatment.
Among the three types of the RAS gene family (HRAS, NRAS, and KRAS), KRAS mutations are most commonly found in colorectal cancer. GTPase, including RAS, normally hydrolyze GTP into GDP to maintain inactivity; however, when KRAS is mutated, GTPase activity is blocked, thus continually activating RAS. Most mutations occur at codons 12 and 13 and, in rare cases, at codon 61.
KRAS testing is used to establish the proper treatment continuum by rapidly checking the KRAS state in early stages rather than selecting the preferred regimen. Because anti-EGFR drugs are not used to treat patients with colorectal cancer in stages 1–3, the KRAS gene test is also not recommended for these patient groups. For KRAS wild-type metastatic colorectal cancer patients, adding anti-EGFR antibody to the anticancer treatment results in a higher overall response rate and a longer overall survival period compared with the anticancer drug-only treatment [29]. Although there is some controversy that codon 13 mutations (p.G13D) cannot predict treatment non-response [8990], these are hypothetical and require further prospective studies. Therefore, the use of anti-EGFR drugs is not currently accepted as a conventional treatment for patients with p.G13D mutations. Additionally, since there are some cases where anti-EGFR drugs show no effect on patients with wild-type KRAS, the issue of whether there is an appropriate additional biological indicator for cetuximab or panitumumab sensitivity other than KRAS testing is being studied. The frequency of KRAS codon 12 and 13 mutations in colorectal cancer patients is 39.3%, and the most common mutations are p.G12D (36%) and p.G12V (21.8%), both in codon 12 [91]. Ethnic differences in KRAS mutation frequency have not been established.
KRAS mutation testing is performed by using specimens obtained from surgery (primary or metastatic colorectal cancer tissue), endoscopic biopsy, or thin-needle aspiration samples; this can be done by using frozen or paraffin-embedded tissue. There is no established standard for determining the amount of tumor cells within the specimen that is necessary for the KRAS gene test; however, as testing technology advances, the required tumor cell amount is decreasing. It is desirable to use at least six to 10 tissue pieces that are 5 µm in size, and when possible, it is also desirable to use a specimen with high tumor cell amount.
KRAS mutation occurs in early stages of colorectal cancer formation, so there is no large difference in the mutation state of the primary cancer tissue and the metastatic cancer tissue. Therefore, the KRAS gene testing can be performed by using either the stored primary cancer tissue or the metastatic cancer tissue. Tissue biopsy should not be conducted only to perform the KRAS gene test unless there is neither primary cancer tissue nor metastatic cancer tissue.
There are many methods that can be used for KRAS gene test, and each method has its pros and cons. For this reason, no specific testing method is recommended for KRAS mutation testing. Direct sequence testing can detect all mutations existing in the tested area. This method has high specificity but low sensitivity (sensitivity of about 10–25%), so a large quantity of DNA containing the mutation (at least 50% of tumor cells) is needed. Pyrosequencing can detect cancer, if 5–10% of the entire DNA has the mutation. Real-time quantitative PCR can detect only specific target mutations, and the sensitivity is 1–10% depending on the technology (TaqMan or Scorpions ARMS, etc.). When PCR is combined with mutant enrichment technology, the sensitivity can be improved to 0.1% or less [28].
If a mutation is detected, the tumor is classified as unresponsive to cetuximab and panitumumab; if no mutation is detected, it is classified as responsive to cetuximab and panitumumab [27].


 XML Download
XML Download