

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory disorder that is characterized by fever, hepatosplenomegaly, and cytopenia. Primary HLH is an autosomal recessive disease and appears in infants or young children, whereas secondary HLH affects older children or adults who do not have a family history of similar conditions or a confirmed genetic disorder. Known gene mutations and their associated familial HLH (FHL) subtypes are as follows: PRF1 with FHL2, UNC13D with FHL3, STX11 with FHL4, and most recently, STXBP2 with FHL5 [1]. Here, we report a rare case of HLH with a novel PRF1 mutation.
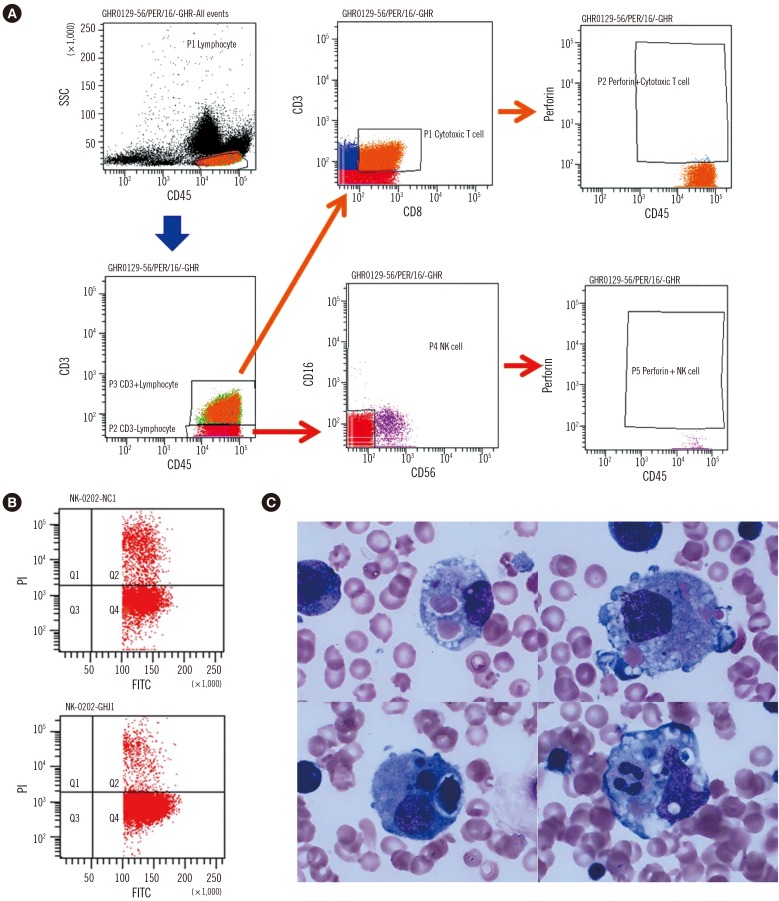
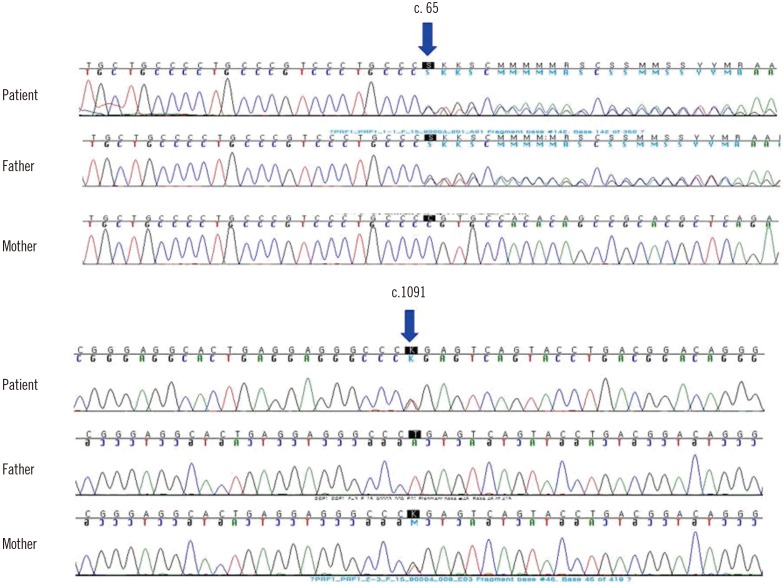
A two-month-old female was admitted because of a one-week history of fever and cytopenia that did not respond to antipyretics or empirical antibiotic therapy. She was born at 39+6 weeks of gestation with a birth weight of 3.5 kg. She had no siblings, and her family history was unremarkable. At admission, the patient's temperature was greater than 38.5℃ and splenomegaly was present (>3 fingers). Laboratory examinations revealed bicytopenia (hemoglobin 66 g/L, platelets 20×109/L, and leukocytes 11.2×109/L); hypofibrinogenemia (42 mg/dL, reference interval [RI]: 200–400 mg/dL); and elevated levels of ferritin (9,542.7 ng/mL, RI: 10–290 ng/mL), soluble CD25 (21,800 U/mL, RI: 122–496 U/mL), AST (565 IU/L, RI: 0–40 IU/L), ALT (283 IU/L, RI: 0–40 IU/L), and lactate dehydrogenase (600 IU/L, RI: 120–250 IU/L). Flow cytometry, which was performed as previously reported [2], revealed decreased intracellular perforin expression on the patient's natural killer (NK) cells compared with levels observed in a healthy individual (Fig. 1A). Flow cytometry [3] also revealed markedly reduced NK cell cytotoxic activity compared with activity observed in a healthy individual (Fig. 1B). A bone marrow examination, which was performed immediately after admission, showed active hemophagocytosis (Fig. 1C). Serologic tests were compatible with a remote infection of Epstein-Barr virus (EBV) or cytomegalovirus (CMV). Complete sequencing of the PRF1 gene revealed the following compound heterozygous mutations: c.65delC (p.Pro22Argfs*29) and c.1091T>G (p.Leu364Arg). Her father was heterozygous for the former mutation, and her mother was heterozygous for the latter (Fig. 2). The Leu364Arg substitution was considered to be a probably damaging variant (score: 0.978) by PolyPhen-2 analysis (http://genetics.bwh.harvard.edu/pph2/) and a disease-causing variant (probability: 0.895) by MutationTaster analysis (http://www.mutationtaster.org). Disease-associated variants were not detected in UNC13D. On the basis of these findings and established criteria, the patient was diagnosed as having HLH, specifically FHL2 [1]. After receiving treatment based on the HLH-2004 protocol, which included dexamethasone, etoposide, and cyclosporine A [4], the patient showed clinical and laboratory improvement. However, eight months later, the patient was found to have reactivation involving the central nervous system, which was treated by intrathecal methotrexate and repeated chemotherapy. Upon recovery, she underwent haploidentical hematopoietic cell transplantation (HCT) 12 months after her initial presentation. At the time of this publication, the patient had survived for 10 months post-transplantation, although she developed HCT-associated complications including reactivation of EBV and CMV infections.
The types of PRF1 mutations in Korean patients with FHL are notable for two characteristics. First, the frequency of PRF1 mutations was 4.2% among Korean patients in a recent study [5], whereas these mutations account for approximately 30–35% of FHL cases in other ethnic groups [1]. These low frequencies are accounted for by the unexpected predominance of UNC13D mutations (85% in a recent study) in Korean patients with FHL [5]. Second, a specific mutation, c.1090_1091delCT, is the most prevalent PRF1 mutation in Korean and Japanese patients, compared with that in other ethnic groups [56789]. Interestingly, this type of PRF1 mutation shares the same nucleotide region as the mutation observed in our patient (c.1091T>G). c.65delC is the only other mutation reported in Korean FHL2 patients [57]. Our patient had compound heterozygous mutations in the PRF1 gene. The c.1091T>G mutation has neither been previously reported as a single nucleotide polymorphism (SNP) nor as a mutation in the main genomic databases including the Human Gene Mutation Database, the SNP database (dbSNP), and ClinVar, whereas the c.65delC mutation was previously reported in Asian infants in a compound heterozygous manner [710].
In summary, we report a patient with typical HLH due to compound heterozygous mutations in PRF1, which manifested ascytotoxic lymphocyte dysfunction. To the best of our knowledge, this is the first case report worldwide of a patient with the PRF1 mutation c.1091T>G. This mutation shares the same site as the mutation c.1090_1091delCT, which is a unique PRF1 mutation in the Korean and Japanese populations.
 XML Download
XML Download