

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most prevalent X-linked enzymopathy. G6PD is the first enzyme in the pentose phosphate pathway, and NADPH generated by the pathway provides an important source for intracellular reduction, particularly for red blood cells (RBCs) [1]. Since G6PD is the only NADPH-producing enzyme in RBCs, its activity in these cells provides defense against oxidative damage. Acute hemolytic anemia is a common clinical symptom of the deficiency, but G6PD-deficient individuals usually have no clinical manifestations and remain asymptomatic until they are exposed to a hemolytic trigger. The triggers include various exogenous agents, such as infection and hemolysis-inducing drugs, and can each cause jaundice, hyperbilirubinemia, and hemoglobinuria. When a G6PD deficiency is suspected, a patient receives various tests, including a complete blood count (CBC) with reticulocyte count, direct and indirect bilirubin levels, lactate dehydrogenase (LDH), Coombs test, and G6PD enzyme activity. A genetic analysis by G6PD sequencing is also available.
According to the WHO classification, G6PD deficiency is divided into five classes on the basis of the severity of the enzyme deficiency as measured by the level of RBC G6PD activity and clinical manifestations [2]. The majority of patients with G6PD deficiency belong to Class II, characterized by a severe enzyme deficiency, but rare G6PD-deficient individuals fall into Class I, with an even more severe enzyme deficiency related to chronic non-spherocytic hemolytic anemia (CNSHA). Genetic diagnostic methods can be used to identify asymptomatic patients who are not in an acute aggravation state, even those with a Class IV G6PD deficiency, with enzyme activity levels within the normal, reference range, but who have the potential for aggravation in response to triggers.
Since G6PD Riley and Guadalajara were first reported by our institute [34], two additional G6PD deficiency patients have been genetically confirmed in Korea [56]. We described three more Korean cases of genetically confirmed G6PD deficiency, covering the laboratory profiles of all seven patients including previously reported cases, and investigated mutations in G6PD using an in silico approach. We also compared the simulated effects of the G6PD mutations to WHO classes according to the level of enzyme activity in RBCs and clinical manifestations.
METHODS
1. Patients
All seven known Korean male patients with G6PD mutations including four previously reported cases were examined. The seven patients experienced episodes of acute aggravation of hemolytic anemia with decreased G6PD enzyme activity. Among them, three patients were newly diagnosed as G6PD-deficient in this study. The G6PD enzyme activity levels in the RBCs of all three patients were low, i.e., 10.5, 2.1, and 0.8, respectively (reference range for men: 7.9–16.3 U/g Hb). The study protocol was approved by the Institutional Review Board of The Catholic University of Korea, and written informed consent for clinical and molecular analyses was obtained from the three newly diagnosed cases.
2. Biochemical analysis of G6PD enzyme activity levels
A spectrophotometric assay was used to quantify G6PD enzyme activity (Ben S.r.l. Biochemical Enterprise, Milan, Italy) by measuring the formation of NADPH molecules (based on absorbance at 340 nm). Fluorescence was detected by using a Hitachi U-3010 UV-Visible, Scanning Spectrophotometer (Hitachi, Tokyo, Japan).
3. Direct sequencing
A genetic analysis was performed by direct sequencing of the G6PD. Genomic DNA was extracted from peripheral blood by using a QIAamp DNA Mini Kit (Qiagen GmbH, Hilden, Germany). Entire coding exons and flanking intronic sequences of G6PD were amplified by PCR using different combinations of 11 primer sets designed using Primer3 (http://bioinfo.ut.ee/primer3/) by the authors. Direct sequencing of PCR products was performed by using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA), and the products were resolved on the ABI 3130XL Genetic Analyzer (Applied Biosystems). Sequence electropherograms were analyzed by using Sequencher 4.9 (Gene Codes, Ann Arbor, MI, USA). The G6PD sequence with RefSeq ID NM_001042351.2 was used as a reference for cDNA nucleotide numbering. All identified variants were confirmed by bidirectional resequencing.
4. In silico analysis of identified amino acid residues
1) Sequence variant databases
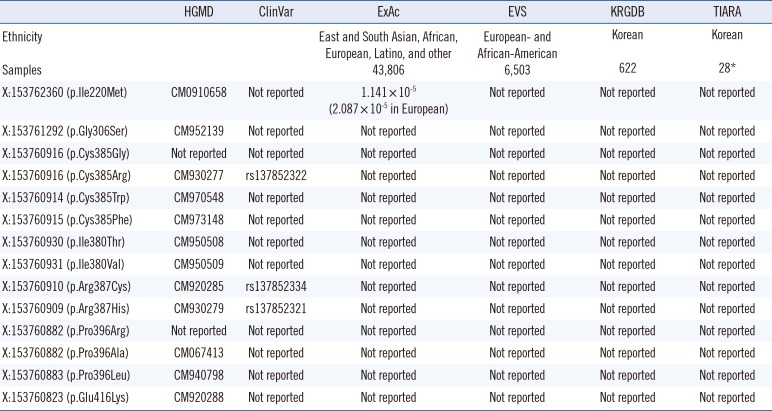
Other G6PD mutations at the identified amino acid residues were retrieved from the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/) [7], ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) [8], and the 1000 Genomes Project (http://browser.1000 genomes.org/) [9]. The Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) was also searched; this source provides exome sequencing data from a variety of large-scale sequencing projects, including 60,706 unrelated individuals, and serves as a useful reference set of allele frequencies for severe disease studies. An allele frequency of less than 0.001 indicates that a variant is rare in the healthy population and the variant can be interpreted as a pathogenic mutation. The Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS/), which includes 6,503 samples drawn from multiple ESP (Exome Sequencing Project) cohorts, representing all ESP exome variant data, the Korean Reference Genome DataBase (KRGDB, http://152.99.75.168/KRGDB/menuPages/firstInfo.jsp/), which harbors whole genome sequencing data for 622 Korean individuals, and the Total Integrated Archive of short-Read and Array (TIARA, http://tiara.gmi.ac.kr/) database, including data generated by next-generation sequencing and high-resolution comparative genomic hybridization arrays, were also searched.
2) Computational prediction tools
There are many prediction tools for the effects of genetic variants that use various methods for prediction. These tools provide useful information, but sometimes the predictions for the same variant do not agree. Thus, several prediction tools were compared to evaluate the mutations identified in Korean patients. On the basis of this comparative analysis, the most suitable method for predicting the effects of G6PD mutations was identified. The following widely established computational prediction methods were applied: the evolution-based prediction tools including Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org/) [10], the Protein Variation Effect Analyzer (PROVEAN, http://provean.jcvi.org/) [11], Multivariate Analysis of Protein Polymorphisms (MAPP, http://mendel.stanford.edu/SidowLab/downloads/MAPP/) [12], and Align-GVGD (http://agvgd.iarc.fr/agvgd_input.php) [13]. Protein structure and function were assessed by using straightforward comparative physical and evolutionary analyses and sequence- and structure-based bioinformatics tools, such as Polymorphism Phenotyping v2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/) [14], MutPred (http://mutpred.mutdb.org/) [15], and SNPeffect 4.0 (http://snpeffect.switchlab.org/) [16].
3) Evolutionary conservation analysis
To assess the extent of conservation of the G6PD amino acid residues thought to be associated with disease, the deduced amino acid sequences were compared by aligning the protein sequences of various vertebrate species obtained from the Evolutionary Annotation Database (Evola, http://www.h-invitational.jp/evola/) and conservation was then quantified by using Scorecons [17]. The Scorecons analysis generates a composite score that accounts for amino acid frequencies, stereochemical diversity, gap penalties, and sequence weighting (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/valdar/scorecons_server.pl). The overall diversity of all position scores for the G6PD protein sequence was 0.529, ranging from 0 for non-conserved residues to 1 for highly conserved residues. A G6PD multiple sequence alignment was constructed using the following taxa, which represent broad phylogenetic diversity: human (Homo sapiens; HIT000029531), orangutan (Pongo; ENSPPYT00000024358), macaque (Macaca; XM_001095273), mouse (Mus; BC075663), rat (Rattus; BC081820), horse (Equus; XM_001492232), opossum (Monodelphis; XM_001362790), zebrafish (Danio; XM_694076), medaka (Oryzias; ENSORLT00000015543), and fugu (Takifugu; SINFRUT00000143893).
RESULTS
1. Genetic and phenotypic characteristics of G6PD deficiencies in Korean patients
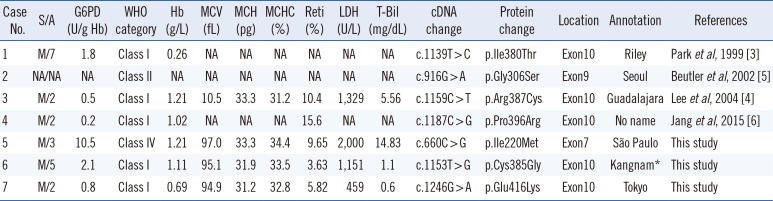
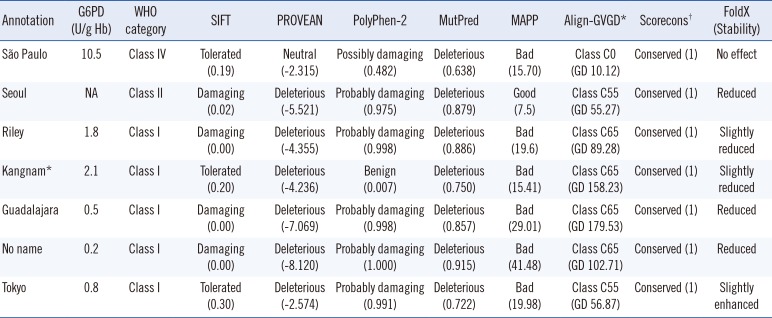
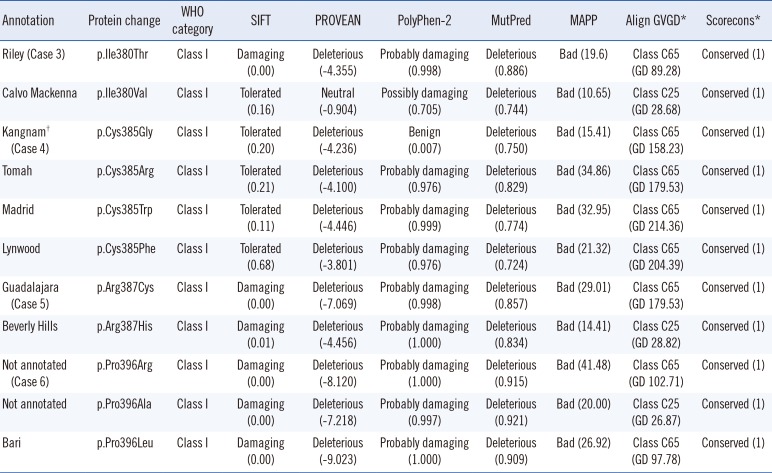
Seven missense mutations were identified as hemizygous (Table 1) in male patients with G6PD deficiencies. Among the seven mutations, five were identified at our institute; one novel mutation (p.Cys385Gly named as G6PD Kangnam) and two known mutations (p.Ile220Met [G6PD São Paulo] [18] and p.Glu416Lys [G6PD Tokyo] [19]) were newly identified in the Korean population in this study. Two known mutations (p.Arg387Cys [G6PD Guadalajara] [20] and p.Ile380Thr [G6PD Riley] [21]) were reported previously by other researchers [34] at our institute. In addition to these five mutations, we found two missense mutations (p.Gly306Ser [G6PD Seoul] [5] and p.Pro396Arg [unnamed] [6]) in a literature review. Mutations affecting the same residue (Cys385) as that of G6PD Kangnam, but resulting in different amino acid changes, p.Cys385Arg (G6PD Tomah) [22], p.Cys385Trp (G6PD Madrid) [23], and p.Cys385Phe (Lynwood) [24], have been described in different ethnic groups.
The three newly diagnosed patients as having G6PD deficiency were characterized. Patient of case 5 was a 3-yr-old boy who suffered from fever and jaundice. He had no family history of hematologic disorders. CBC showed the following: Hb 0.96 g/L, mean corpuscular volume (MCV) 101.3 fL, mean corpuscular Hb (MCH) 32.3 pg, mean corpuscular Hb concentration (MCHC) 31.9%, reticulocyte 17.18%, and undetectable G6PD enzyme activity. The Coombs test and osmotic fragility test were negative. After one week, he recovered from the acute aggravation. The G6PD enzyme level increased to 10.5 U/g Hb, and the G6PD deficiency was categorized as Class IV according to the WHO classification. Sanger sequencing of the G6PD revealed a c.660C>G (p.Ile220Met) mutation, which was previously reported as G6PD São Paulo [18].
Patient of case 6 was a 5-yr-old boy who suffered from a fever and a pale appearance. He had no family history of hematologic disorders. He visited our hospital after recovery from the acute aggravation. The Coombs test and the osmotic fragility test were negative. The G6PD enzyme level was 2.1 U/g Hb without acute aggravation. His condition was classified as Class I G6PD deficiency according to the WHO classification owing to the severely decreased enzyme activity with CNSHA. Sanger sequencing revealed a c.1153T>G (p.Cys385Gly) mutation in G6PD. This mutation was not reported previously; therefore, we named the mutation G6PD Kangnam. Interestingly, mutations affecting the same residue as that of G6PD Kangnam, but resulting in different amino acid changes, p.Cys385Arg (G6PD Tomah) [22], p.Cys385Trp (G6PD Madrid) [23], and p.Cys385Phe (G6PD Lynwood) [24], have been previously described in different ethnic groups.
Patient of case 7 was a 2-yr-old boy who had a fever and a pale appearance. He had a family history of anemia (maternal uncle and younger brother). With acute aggravation, the G6PD level was 0.8 U/g Hb. His condition was classified as Class I G6PD deficiency according to the WHO classification. Sanger sequencing revealed a c.1246G>A (p.Glu416Lys) mutation, which is known as G6PD Tokyo [19].
Four additional mutations were previously reported in Korean individuals. A case study of patient 2, with the G6PD Seoul mutation, was not published, but was described in a later study [5]. The mutation was c.916G>A in exon 9 and a patient with the same mutation was classified as Class II in a later report (the G6PD activity level was 1.6 U/g Hb and there was no CNSHA). Patient 1 was a 7-yr-old boy who presented with mild jaundice and dyspnea. He had neonatal jaundice and recurrent nonspherocytic hemolytic anemia with type B hepatitis. CBC showed an Hb of 0.26 g/L and G6PD activity of 1.8 U/g Hb. On the basis of CNSHA, the patient was Class I according to the WHO classification. He had three brothers who died from severe neonatal jaundice and acute hemolytic anemia. The mutation was c.1139T>C on exon 10 of G6PD. Patient 3 was a 22-month-old boy who presented with jaundice and anemia. He had no family history of hematologic disorders. CBC results were as follows: Hb 0.62 g/L, MCH 33.3 pg, MCHC 31.2%, MCV 106.6 fL and reticulocyte 10.4%. A peripheral blood smear revealed anisocytosis, poikilocytosis, and macrocytosis. Total bilirubin was 5.56 mg/dL, direct bilirubin was 0.57 mg/dL, LDH was 1,329 U/L, and G6PD activity was 0.5 U/g Hb. The patient belonged to Class I according to the WHO classification. Based on sequencing, the mutation was c.1159C>T in exon 10 of G6PD. Patient 4 was a 20-month-old boy who presented with chronic hemolytic anemia and several episodes of acute exacerbation. His Hb level was 1.02 g/L, and G6PD enzyme level was 0.2 U/g Hb. The patient belonged to Class I according to the WHO classification. The mutation was documented as c.1187C>G in exon 10.
2. Effect of mutations on disease manifestation
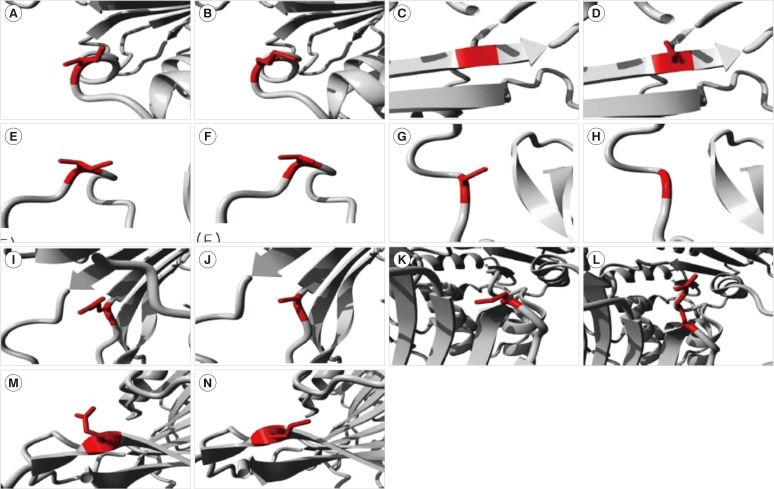
In an in silico analysis, G6PD mutations associated with Class I or II were predicted to be highly deleterious on the basis of their effects on protein structure and function, whereas the effects of Class IV mutations were unclear. Scorecons, an evolutionary conservation predictor, showed that the residues were located in highly conserved regions (Table 2). In terms of evolution-based sequence diversity, the overall diversity based on all position scores for the G6PD protein sequence was 0.529, ranging from 0 for non-conserved residues to 1 for highly conserved residues. Mutations affecting the same residue, such as p.Ile380, p.Cys385, p.Arg387, and p.Pro396, were predicted to be pathogenic on the basis of codon changes (Table 3). FoldX predicted alterations in protein stability, except for G6PD São Paulo, which has been reported in the ExAC database at an extremely low frequency, i.e., 1.141×10-5 (Fig. 1A and B, Table 4). For the Class IV mutation, G6PD São Paulo, an in silico analysis using SIFT, PROVEAN, PolyPhen-2, Align-GVGD, and the FoldX implemented in SNPeffect 4.0 predicted a relatively mild effect, consistent with the observed enzyme activity levels.
DISCUSSION
G6PD deficiencies selectively affect RBCs via two mechanisms. First, most known mutations decrease enzyme stability. Since these cells do not have the ability to synthesize proteins and replenish their enzyme levels, the enzyme level decreases as cells age during their 120-day lifespan in circulation. Second, G6PD activity is decreased, and the diminished ability of RBCs to withstand stress increases the risk of destruction by hemolysis.
To date, approximately 186 G6PD mutations have been documented, most of which (85%, 159/186) are single nucleotide substitutions leading to missense variants [25]. This and previous studies have revealed identical G6PD mutations in different countries around the world. Since 1999, when G6PD Riley was reported at our institute, seven G6PD mutation types have been identified in Korea. The same mutations have also been found in Brazil (G6PD São Paulo) [18], Poland (G6PD Seoul) [26], Americas (G6PD Riley) [21], Mexico (G6PD Guadalajara) [20], and Japan (G6PD Tokyo) [19]. In addition, several mutations can occur at the same nucleotide position, but result in different amino acid changes in different ethnic populations. For example, p.Cys385 variants have been reported as G6PD Kangnam as well as G6PD Tomah [22], Madrid [23], and Lynwood [24]. In addition, p.Pro396 variants have been described as p.Pro396Arg in Korean individuals [6], p.Pro396Ala in Indian individuals [27], and p.Pro396Leu in Italian individuals [28]. These findings and previous studies imply that the same mutations may have arisen by independent mutation events in Korea, rather than being inherited from common ancestral mutations [1819202126].
All of the G6PD mutations affected coding regions, and none has been described in a regulatory region. This suggests that reduced enzyme activity levels are associated with decreased enzyme activity in the G6PD-deficient phenotype, rather than deficiencies in gene expression [2529]. With respect to genotype–phenotype associations, the frequencies of Class I mutations found in exon 10 are significantly higher than those in other exons in G6PD deficiency cases [25]. Mutations located in this region, which encodes the binding interface between the subunits, have a highly deleterious effect on enzyme activity by disrupting the quaternary structure and stability of the protein, and appear to be the most easily identifiable in the general population [1].
In parallel with a genetic analysis, the pathogenicity of each G6PD mutation detected in the Korean patients was predicted by using an in silico analysis according to WHO classes. Class I mutations were predicted to be highly deleterious, while the effects of one Class IV mutation were equivocal. However, all mutations in Korean patients with G6PD deficiencies were located in residues that were conserved across various species. A high degree of evolutionary conservation of certain gene regions is essential for enzyme function and cell survival. Similar to a previous study [18], our G6PD São Paulo mutation with G6PD activity of 10.5 U/g Hb was categorized as Class IV, near the dimer interface. Rare Class IV mutations [518303132] showing nearly normal G6PD activity can be overlooked because molecular analyses of G6PD are only considered when episodes of hemolytic crisis occur.
In summary, identical G6PD mutations associated with G6PD deficiencies in Korean patients may have arisen by independent mutation events. An in silico analysis provided insight into the roles of G6PD mutations from evolutionary and structure-based computational points of view. Further investigations involving more Koreans with G6PD deficiencies are needed to clarify the relationship between the clinical manifestations and mutational spectrum related to this enzyme deficiency.
 XML Download
XML Download