

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Over two decades of scientific research on the HRX/HTRX1/ALL-1/mixed-lineage leukemia (MLL)1 gene-now renamed according to its cellular function into KMT2A-provides an enormous resource of detailed knowledge, but also many questions that are yet unanswered. Throughout this review, I would like to stick to the gene name "MLL" for two reasons: (1) we cloned this gene 20 yr ago [1] and (2) in honor of Prof. Janet D. Rowley who introduced the name "MLL" to the scientific community [2].
Experiments performed in different labs conclusively demonstrated that the expression of various chimeric MLL fusion alleles is sufficient and necessary to drive the onset of leukemia [3456789]. It is presumably the only genetic mutation required for disease onset [10]. This is due to the fact that the wildtype MLL protein assembles into a complex that has a fundamental role in normal cell physiology: this complex-in cooperation with transcription factors-is marking promoters in a cell-type specific manner for gene transcription, thereby creating a 'transcriptional memory system' which is necessary to maintain "lineage identity" in a mitotically stable manner. The MLL complex is also required for embryonic and adult hematopoietic stem cell maintenance [11] and is necessary during embryonal development.
MLL fusion proteins that derive from such illegitimate genetic rearrangements are disturbing these subtle mechanisms and are leading to the onset and maintenance of leukemic stem and tumor cells [12131415].
While many different chromosomal translocations are known to be associated with specific tumor subsets (e.g. PML-RARA with AML FAB M3; BCR-ABL1 with either CML or ALL), the real challenge in MLL-rearranged (MLL-r) leukemia derives from the huge amount of direct (n=82) and reciprocal MLL fusion alleles (n=120) [16], because it raises the question about the pathological mechanisms and/or signaling pathways that are responsible to trigger the conversion of normal hematopoietic stem/progentitor cells into malignant cells. All the yet diagnosed MLL rearrangements are causing similar disease phenotypes (ALL, AML, or MLL), are hard to cure, and display a poor outcome. The only yet existing exception is t(1;11) translocation that expresses the MLL-AF1Q/MLLT11 fusion protein. The presence of this particular MLL fusion protein displays a very good survival of about 90%, indicating that it has only poor oncogenic potential [17].
This review is not trying to recapitulate the already proposed pathomechanisms (HOXA/MEIS1 genes, DOT1L and extended H3K79me2/3 signatures) [81819], but proposes a novel hypothesis to explain the oncogenic properties deriving from the many different MLL fusion proteins. This will help to focus on new research areas in case when currently tested strategies to cure this leukemia subtype will not hold their promises.
CANCER IS CAUSED BY GENETIC MUTATIONS AND EPIGENETIC CHANGES
The development of different cancer types in humans is strictly based on somatic mutations and epigenetic changes. These are still the basic principles after 30 yr of cancer research, and our current knowledge is obtained by next generation sequencing (NGS)-mediated cancer genome sequencing. However, one should be aware that every healthy individual already deviates from the reference genome sequence by roughly 10,000 non-synonymous single nucleotide polymorphisms (SNPs). This large amount of genetic differences is linked to 200-300 loss-of-function mutations and 50-100 gene variations known to be associated with heritable human diseases [20]. These mutations include indels (small insertions or deletions), splice site mutations, and pre-mature stop codons.
Apart from this 'normal' genetic background of healthy individuals, cancer genomes are characterized by additional cancer-type-specific gene mutations and/or gross chromosomal changes, including interstitial chromosomal deletions, chromosomal inversions, or-as in most cases-specific chromosomal translocations. The latter term describes a genetic process where chromatin fragments of two non-homologous chromosomes are being exchanged through an Non-Homologous End Joining (NHEJ)-mediated DNA repair process [2122]. This leads to the creation of "derivative chromosomes" with chimeric fusion genes at the chromosomal fusion sites. Another mechanism that creates "chimeric fusion genes" was identified in solid tumors and was termed "chromothripsis" [23]. Chromothripsis is based on a mistake during mitosis which leads to the fragmentation of a single chromosome due to a micronucleus that forms in a single cell. The chromosomal fragments are then repaired by a random ligation process. This generates a large variety of different chimeric gene fusions; however, most of them are "non-functional" because of out-of-frame, head-to-head, or tail-to-tail gene fusions. Generally, functional fusion genes with oncogenic potential as a result of this "chromatin fragments fusion process" are rare.
FUNCTIONAL CONSEQUENCES OF GENE OR CHROMOSOMAL MUTATIONS
All the above described genetic mutations result in either 'loss-of-function' or 'gain-of-function' situations. Point mutations are mostly associated with functional changes, e.g. changing enzyme activity, changing DNA binding capacity, or changing protein binding capacity. Splice mutations lead to exon skipping and result in altered proteins with arbitrary functions. Frameshift mutations often cause a C-terminal truncation of proteins or mediate an RNA decay mechanism. Premature termination of transcription at cryptic poly A sites may also lead to truncated proteins, but more often lead to an altered protein abundance due to the loss of microRNA binding site which are usually localized in the 3-UTR to control the abundance of proteins.
Balanced chromosomal translocations can be subdivided into two different categories according to their clinical behavior and the resulting cancer mechanism. Category 1 chromosomal translocations are created by the juxtaposition of a cell-type specific enhancer near to a germline gene, which is subsequently strongly transcribed and overexpressed. This is a well described scenario in case of immunoglobuline or T-cell receptor rearrangements, which are specifically associated with lymphoid malignancies, mostly with lymphoma disease phenotypes (e.g. BCL2 or MYC in t(14;18) and t(8;14) translocations, associated with Follicular and Burkitt's lymphoma). Category 2 translocations recapitulate an important evolutionary principle, namely "exon shuffling". The latter process has been successfully used throughout evolution to generate new genes with novel properties. Category 2 chromosomal translocations recapitulate this evolutionary process by recombining two genes from different chromosomes. The resulting chimeric genes are transcribed into "fusion mRNAs" and translated into "fusion proteins" with oncogenic potential. The presence of specific fusion genes is always associated with particular cancer types. Again, the presence of such fusion proteins often results in 'loss-of-function' (normal properties) and 'gain-of-function' (oncogenic properties) situations. Many of these fusion proteins have already been tested in functional assays and/or in mouse models (see above), solely to demonstrate that they have indeed the capacity to induce a malignant transformation in normal cells that carry otherwise no known mutation. Therefore, these cancer-specific fusion proteins ("oncofusion proteins") are the primary targets for scientific research and drug development.
CHROMOSOMAL TRANSLOCATIONS, THE MLL GENE AND ACUTE LEUKEMIA
So far, a total of 572 human genes have been identified to be involved in cancer (Cancer census database [http://cancer.sanger.ac.uk/cosmic/census] of the Sanger Institute; dated September 2015). Of those, 354 genes were identified in chromosomal translocations that are recurrently diagnosed in different human cancers. In hematological neoplasias, chromosomal translocations are the hallmark for acute leukemias (ALL or AML). Acute leukemias are frequently associated with specific gene fusions. A particular group of patients is characterized by so-called "MLL fusions". They represent about 5-10% of all acute leukemia cases in childhood and adult leukemia. MLL fusions are based on genetic rearrangements of the MLL gene. Until today, about 80 direct (MLL-X) and 120 reciprocal MLL fusion genes (X-MLL) have been described in acute leukemia patients (see [16]: Supplemental Table S4).
The cDNA of human MLL has been cloned 23 yr ago in four different labs. First, the HTRX1/MLL cDNA was shown to span a gene localized at 11q23, and chromosomal breakpoints in 11q23-leukemia patients are disrupting this gene [224]. Subsequently, two other groups cloned successfully the first MLL fusion cDNAs (HRX/ENL and ALL-1/AF4) [2526]. Four years later, the genomic MLL gene structure has been unravelled by cloning [127]. To our knowledge, the MLL gene exhibits 37 exons, but the 99 nucleotide long exon 2 is spliced out in about 66% of all transcripts [28]. Therefore, most scientists-but also public databases-are tending to display the MLL gene only with 36 exons. The correct nomenclature should list 37 exons for MLL, because this defines the major breakpoint cluster region localizing between MLL exons 9 to 14; this is a nomeclature which is used by nearly all researchers.
Different transcripts of the MLL gene give rise to a ~500 KDa protein. If being very accurate, the MLL protein is coming in eight different flavors with either 3,958, 3,961, 3,969, and 3,972 or 3,991, 3,994, 4,002, and 4,005 amino acids. This is due to the skipping of MLL exon 2 (encoding 33 amino acids) and four alternative splice events that occur at the border between MLL exon 15 and 16. The splice variant at MLL exon 15 and 16 gives rise to particular changes in the ePHD3 domain with important consequences for MLL protein functions [29].
The MLL protein is quite important to sustain normal cell physiology. Vice versa, any type of genetic disruption in combination with the expression of MLL fusion alleles seems to orchestrate a situation which is associated with epigenetic changes, deregulated gene transcription, and the aquirement of stem cell-like features, finally leading to a malignant transformation of the affected cell.
ACUTE LEUKEMIA IS CAUSED BY DIFFERENT MLL FUSION ALLELES
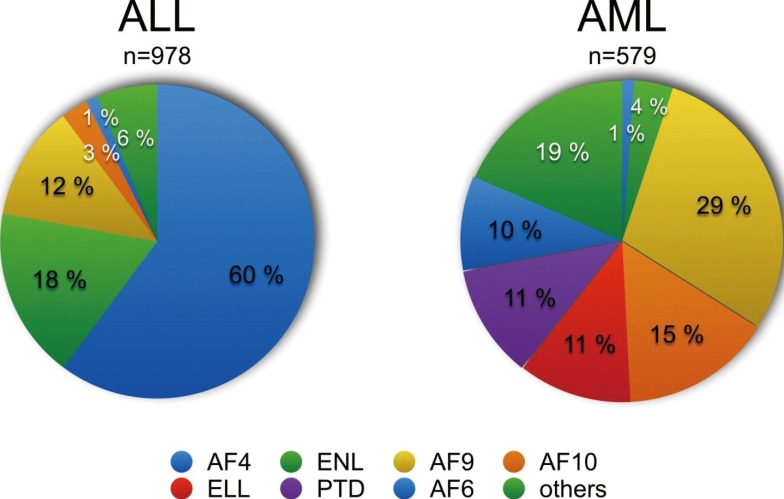
MLL translocations (n=82) can be diagnosed in about 5-10% of all acute leukemia patients. However, the majority of patients are caused by translocations that involve only very few fusion partner genes. If analyzed by disease phenotype, the majority of ALL patients (~90%) are caused by the three gene fusions, namely MLL-AF4/AFF1, MLL-AF9/MLLT3, and MLL-ENL/MLLT1. The gene fusions MLL-AF10/MLLT10 (~3%) or MLL-AF6/MLLT4 (~1%) do not play a significant role in terms of patient numbers (Fig. 1 left panel). This pictures is extending in AML patients where the majority of patients (~76%) are caused by an MLL-AF9, MLL-AF10, MLL-ELL, MLL-AF6 fusion, or a partial tandem duplication (PTD) of the MLL gene (Fig. 1 right panel). This general distribution does not change significantly when breaking down to infants, pediatric, or adult patients with the above mentioned rearrangements [16].
The remaining list of recurrently diagnosed fusion partner genes is long and comprise-besides EPS15/MLLT5, AF1Q/MLLT11, AF17/MLLT6, and different Septin genes-about 70 other genes that have been identified and described (see [16]: Supplemental Table S4). However, in terms of clinical relevance these represent rare cases and are clinically not of importance, but they are quite useful and probably of interest for basic research. All these fusion genes bear the potential to retrieve novel clues about disease mechanisms and target structures.
THE GENERAL PATHOLOGICAL CONCEPT BEHIND MLL TRANSLOCATIONS
By simply looking at this comprehensive list of MLL fusion partners, we have to think about potential disease mechanisms. Actually, we know that some of the known partner proteins (AFF1/AF4, AFF2/LAF4, AFF4/AF5, MLLT3/AF9, MLLT1/ENL, MLLT10/AF10, MLLT6/AF17, and ELL) are involved into the control of 'transcriptional elongation', as they bind either directly to RNA polymerase II (RNAPII) or a part of super elongation complexes which interacts with RNAPII [30313233].
However, we have no clue about the pathological role for most of the other fusion partner genes in the context of their MLL fusion. The list of more than 70 fusion partners encompasses cytosolic enzymes, nuclear proteins, membrane proteins, extracellular matrix protein, mitochondrial enzymes, and cytoskeleton proteins, but none of these proteins give immediately a hint for a disease mechanism.
Therefore, I would like to propose a new hypothesis that could potentially explain the oncogenic mode-of-action provided by many different MLL fusion alleles. The very high number of oncogenic MLL fusion alleles can be explained by two independent mechanisms.
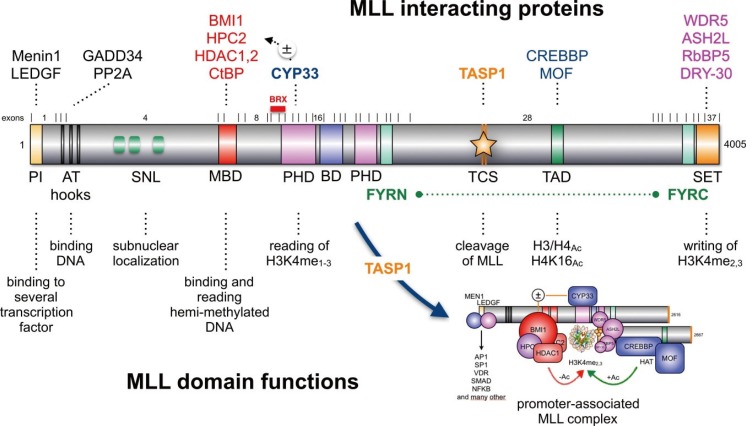
Mechanism 1 can be explained by looking at Fig. 2 to 4. In Fig. 2, the human MLL protein is depicted. The MLL protein represents a multi-binding interface for a large variety of different nuclear proteins and exhibits epigenetic reader and writer functions. The MLL protein is processed by Taspase1 [34], resulting in two protein fragments (p320 and p180) that bind to each other in order to form a molecular hub for the assembly of a large nuclear complex. Described binding proteins are: MEN1 and LEDGF, GADD34, PP2A, the PAF complex, a Polycomb group complex (BMI1, HPC2, HDAC1/2, CtBP), CYP33, CREBP and MOF, and the SET domain core proteins (WDR5, RbBP5, ASH2L, SRY-30) [3536373839404142434445]. The N-terminal portion of the MLL protein (until amino acid 2,616; 1st Taspase1 cleavage site) is functionally linked to bind and read chromatin signatures, while the C-terminal portion of the MLL protein (2nd Taspase1 cleavage site: amino acid 2,667-4,005) is performing enzymatic functions, namely to acetylate and methylate histone core particles. This way, the MLL complex binds to promoter regions of active genes, marks these regions by covalent histone modifications (epigenetic modifications), and establishes thereby a transcriptional memory system that is necessary for lineage identity.
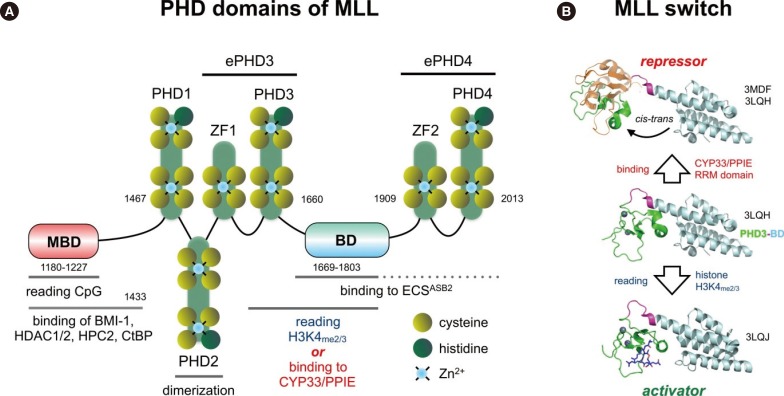
However, there is another important function of MLL, which needs to be explained in more detail to understand the impact of chromosomal translocations. Near the center of the MLL protein is a 'PHD domain'. This region is composed by PHD1-3 subdomain, a bromo domain (BD) and another PHD4 subdomain. The PHD domain exhibits two normal PHD subdomain structures (PHD1/2: Cys3-His-Cys4), while PHD3 and 4 are so-called 'extended PHD subdomains' (ePHD3/4: Cys4---Cys3-His-Cys4) (Fig. 3A). The PHD subdomains 1-3 are followed by a BD, which has in the MLL protein no histone acetyl reading function rather than stabilizing the ePHD3 domain. In particular, the ePHD3 subdomain is required to read H3K4me2/3 signatures within the chromatin [46]. However, when the ePHD3 subdomain binds to CYP33/PPIE, a prolylisomerase, a conformational change is catalyzed that disables the ePHD3 subdomain to interact with the BD domain [4748]. As long as ePHD3 subdomain is docked via a protein helix to BD (see Fig. 3B), it exhibts its essential reader function for nucleosomal H3K4 methylation signatures. Isomerization via CYP33/PPIE allows to disconnect the ePHD3 subdomain from the BD domain and to interact with the BMI1/HPC2/HDAC1-2/CtBP complex that becomes then enabled to bind to the Methyl-DNA binding domain (MBD in Fig. 2 and Fig. 3A). Binding of MLL to this Polycomb-group proteins converts the MLL into a transcriptional repressor. This defines the CYP33/PPIE isomerase as a master switch that triggers the MLL complex between two different modes of action: transcriptional activator or repressor. Nothing is known about the precise details of this molecular switch mechanism, but it is highly likely that it depends on the promoter context and/or signaling pathways. This "MLL switch" is responsible for the known effects on gene transcription: when MLL knock-out cells are transcriptionally profiled together with their isogenic wild-type cells, then more genes become upregulated (66%) than downregulated (33%) in the knock-out situation [49].
Therefore, the complex functions exerted by the MLL protein can be summarized by its ability (see Fig. 4A) to perform binary decisions ("Yes" or "No") for gene transcription. Binding of CYP33/PPIE to the ePHD3 subdomain serves thereby as a molecular trigger to toggle between the two modes of action.
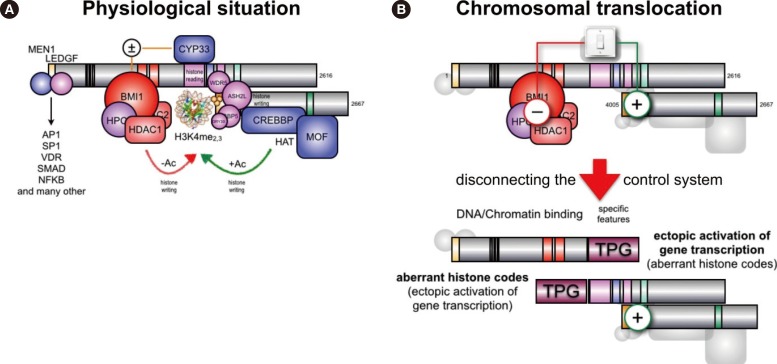
What does actually happen when a chromosomal translocation occurs at the MLL gene? Chromosomal rearrangements ususally separate the MBD from the PHD domain (see Fig. 4B), thereby destroying the above described intrinsic control mechanism of the MLL protein. Even the binding of CYP33 to the PHD domain is impaired, at least when the chromosomal breakpoint localizes within MLL intron 11 [29]. In principle, "chromatin reading" functions become now separated from the "chromatin writing" functions. Consequently, both separated portions of the MLL protein become constitutively active, regardless of their fused protein sequences. The MLL-X fusions still bind via MEN1/LEDGF and the PAF complex to chromatin and associated transcription factors in promoter regions, but are disabled to exert any inhibitory function. The reciprocal X-MLL fusion proteins retain the ePHD3 chromatin reader domain, the CREBBP/MOF binding domain as well as the SET domain complex. However, even when CYP33/PPIE binds to the PHD domain of X-MLL fusions, the binding of the Polycomb (BMI-1/HPC2/HDAC1/2) repressor complex is disabled owing to the missing MBD. This was already nicely demonstrated by experiments, where the PHD domain was artificially fused to existing MLL-X fusion proteins. This was sufficient to eliminate their oncogenic properties, because repressing functions are now exerted by the fused PHD domain via recruiting to the BMI-1 repressor complex [5051].
This is highly similar to other known chromosomal translocations, where other regulatory systems become destroyed as well. As an example, the BCR-ABL1 translocation destroys an instrisinc control mechanism of the ABL kinase and makes it constitutively active in the BCR-ABL1 fusion protein; PML-RARA converts an "activator of gene transcription" into a "repressor of transcription", similar to what happens with the RUNX1-RUNX1T1 (AML1-ETO) fusion protein. Thus, all other yet known translocations act through the destruction of regulatory mechanims. In case of MLL, a regulatory mechanism that controls epigenetic mechanisms becomes destroyed.
BREAKPOINT LOCALIZATION WITHIN THE MLL GENE DEFINES THE OUTCOME OF PATIENTS
Arguments in favor of the above described hypothesis come from the analysis of chromosomal breakpoints in leukemia patients. The breakpoint cluster region usually encompasses the region between MLL exons 9 to 14. In rare cases, AF6 translocations end up in MLL intron 21 or 23; however, these are clear exceptions from the here depicted mechanisms and are moreover exclusively associated with T-ALL [525354].
It has been described that breakpoints within the MLL gene mainly cluster to MLL introns 9-11 [555657], but the breakpoint distribution is also somehow linked to the age of patients: adult leukemia patients (late disease onset) tend to have their breakpoints mostly in MLL intron 9 and 10, while infant ALL patients (early disease onset) have their breakpoints mostly in MLL intron 11 [58]. As already mentioned above, MLL exons 11 to 16 code for the PHD1-3 domain (see Fig. 3). Breakpoints in MLL introns 9 and 10 do not impair the structure of the PHD domain, while breakpoints in MLL intron 11 do so, because two important cystein residues are missing. Since the PHD domains are Cystein-histidine-rich motifs (Cys3-His-Cys4), the missing cysteine residues are preventing a correct folding of the PHD domain, and thus, may impair some of their known functions. According to our own experimements [29], it definitively impairs the dimerization capacity and compromize CYP33 binding. However, it may potentially also influence binding of the degradation protein ECSASB2 [59]. This could be a possible explanation for the extremely long half-life of the AF4-MLL fusion protein [60].
A recent study has demonstrated for the first time that MLL intron 11 breakpoints are associated with a worse clinical outcome [61]. Thus, the physical separation of the MBD from the PHD defines per se the first oncogenic hit, due to the loss of the inhibitory control switch. However, breakpoints within MLL intron 11 may even worsen the situation, because additional features deriving from PHD domain of MLL are compromised as well (e.g. binding to CYP33, degradation pathway, etc.). Thus, breakpoints localizing in MLL intron 11 behave functionally like a "second hit".
THE EPIGENETIC EFFECTS DERIVING FROM MLL FUSION PROTEINS
The second mechanism derives definitively from functions exerted by the N- and C-terminally fused protein sequences. In most cases, these fused protein sequences exhibit accessory functions, e.g. changing the protein interactome. An example for this scenario is fusions with AF4, LAF4, or AF5. They all contain the first 360 amino acids of their cognate wildtype proteins. This portion of AF4/AF5/LAF4 is known as a docking hub for proteins involved in transcriptional elongation control [3362]. A whole series of proteins bind to these tiny protein portions, e.g. P-TEFb, NFkB1, DOT1L, and NSD1 amongst others. This gives those reciprocal fusion proteins some very unique features that are not present in any other known reciprocal MLL fusion. One of these features is the ability to directly bind to and travel together with RNAPII during transcription. This has several consequences, because the associated histone methyltransferases, like DOT1L or NSD1, in conjunction with the SET-domain of MLL is changing the epigenetic imprinting at transcribed gene loci (a combination of H3K4me2/3, H3K36me2/3, and H3K79me2/3 marks). Setting this type of 'histone code' onto transcribed genes with these signatures may convert a normal "gene body signature" into a "promoter signature" which could be one of the reasons for oncogenic conversion (see below and [33]).
According to this model, all MLL target promoters can now be bound by MLL-X fusion proteins, which in turn become strongly enhanced in their transcription activator function. Vice versa, a few reciprocal X-MLL fusion proteins (e.g. AF4-MLL) exert their chromatin modifying functions in a RNAPII-dependent manner, which may result in the above described aberrant H3K4/36/79 methylation signatures. This strange signature may cause a situation where an inactive chromatin region-as a consequence of normal differentiation processes-becomes reactivated and thereby causing a "non-differentiated state". Such a process is usually accompanied by the re-expression of stem cell genes (NANOG, OCT4), which has already been observed in cells expressing t(4;11) fusion proteins [6364]. This type of back-differentiation could be quite similar to what happens when induced pluripotent stem (iPS) cells are generated by expressing a combination of distinct sets of transcription factors [65].
Since all yet tested MLL fusion proteins-expressed in Lineage-, Sca1+ and Kit+ hematopoietic stem/progenitor (LSK) cells and transplanted back into the mouse system-need a minimum of 6-12 months for the development of leukemia, it may argue for a long-term epigenetic process in order to convert a normal cell into a malignant cell. Such an "epigenetic disease mechanism" may not require additional mutations rather than a certain time frame to slowly change the epigenetic layer in affected cells.
THE SYSTEMATIC ANALYSIS OF MLL FUSION PARTNERS: A NEW CLASSIFICATION
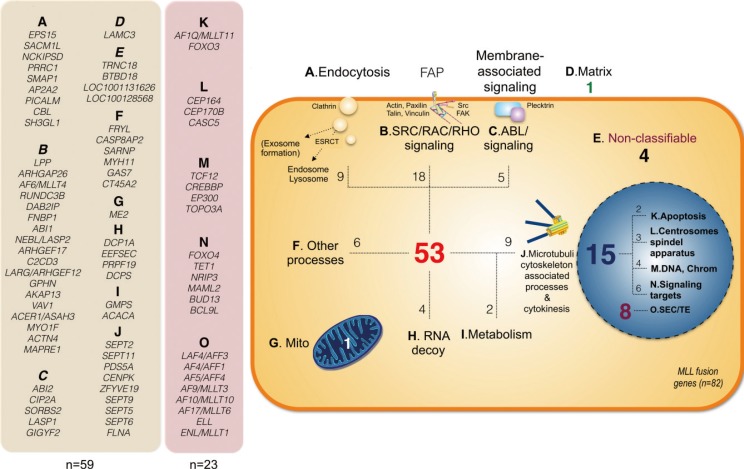
Assuming that the above mentioned hypothesis about the two mechanisms explains the onset of leukemia by the huge number of different MLL fusion alleles, we have to take a closer look to the functional properties that are exerted by the currently known 82 fusion partner proteins (79 have been published so far; see [16]).
When looking to the current list of fusion partner genes, a dozen of them can be immeditely taken from the list, because we know their function(s). These are AFF1/AF4, AFF2/LAF4, AFF4/AF5, MLLT3/AF9, MLLT1/ENL, MLLT10/AF10, MLLT6/AF17, and ELL (Fig. 5). They represent the most frequent fusion genes in terms of diagnosed cases in acute leukemia patients. Interestingly, they are all involved in the process of "transcriptional elongation", which has been intensively investigated in many publications and reviews (e.g. see [6667]). These proteins are either directly a part of "super elongation complexes" (SEC) or directly associated with RNAPII and deregulate its transcriptional activity but also to cause dramatic epigenetic changes.
Four other fusion partner genes (TRNC18, BTBD13, LOC100131626, and LOC100128568) can be taken out from the list, because there is no functional description available in the literature-apart from their fusion to MLL gene.
For the remaining 70 MLL fusion genes, we present here a novel classification system, according to their function and/or their protein interaction profile (Fig. 5). These 70 MLL fusion partner genes can be roughly divided into 53 cytosolic proteins, 15 nuclear proteins, one extracellular matrix protein and an enzyme of the mitochondrial matrix. Interestingly, the 53 cytosolic proteins can be subcategorized into different functional classes: four are involved in RNA decay degradation, two represent normal metabolic enzymes, and six belong to singular cellular processes. All others (n=41) are somehow related to cellular signaling processes: five proteins are involved in ABL kinase or other signaling pathways, nine are directly linked to 'Clathrin-mediated endocytotic processes' and downstream signaling pathways, nine are linked to centrosome-/microtubuli-mediated signaling processes (mitosis and cytokinesis). The majority of 18 proteins are linked to aspects of focal adhesion plaque (FAP) signaling linking to RAC/RHO signaling pathways that influences the cytoskeleton and cell migration. The latter process is also linked to cell cycle control and growth. Therefore, one might speculate that these MLL fusion partners are not completely random but are pin-pointing that certain signaling pathways that need to be impaired.
RHO/RAC signaling, in particular RHO-GEF and RAC-GAP functions, plays an important role in cytoskeleton modeling by regulating the activity of Rho-like GTPases, such as RHO, RAC, and CDC42. Moreover, these signaling pathways have already been related to the onset of lymphoid-commited cancer stem cells in another leukemia entity (ABL-BCR fusion proteins and development of ALL) [686970]. The ABL-BCR fusion proteins (p48 or p96 in case of p235 or p185 BCR-ABL1, respectively) are expressed from the reciprocal fusion alleles in t(9;22) translocations. Both fusion protein variants are able to trigger RAC/RHO/CDC42 signaling events, while only the p96 fusion is able to exert oncogenic functions and is essentially involved in the creation of leukemic stem cells in p185+ ALL. Interestingly, the corresponding p185 BCR-ABL1 fusion protein is unable to do so, but is required for the infinite growth of the tumor bulk. Thus, targeting BCR-ABL1 via tyrosine kinase inhibitors (TKI: Imatinib and derivatives) allows to reduce the tumor bulk, but it needs other therapeutics, like ATO (As2O3), to kill leukemic stem cells [71].
Another interesting paper reported the role of FRAT2 in MLL-rearranged cancer cells [72]. MLL fusion proteins cause an activation of RAC GTPases via FRAT2. FRAT2 activates RAC through a signaling mechanism that requires glycogen synthase kinase 3β (GSK3β) and Dishevelled (DVL), which are both a part of the canonical WNT signaling pathway. Disruption of this pathway abrogates the leukemogenic activity of MLL fusions. This suggests a rationale for the alreday published requirement of the canonical WNT signaling for oncogenic effects of distinct MLL fusions [73].
OTHER CLUES-THE INTERACTOME OF FUSION PARTNERS
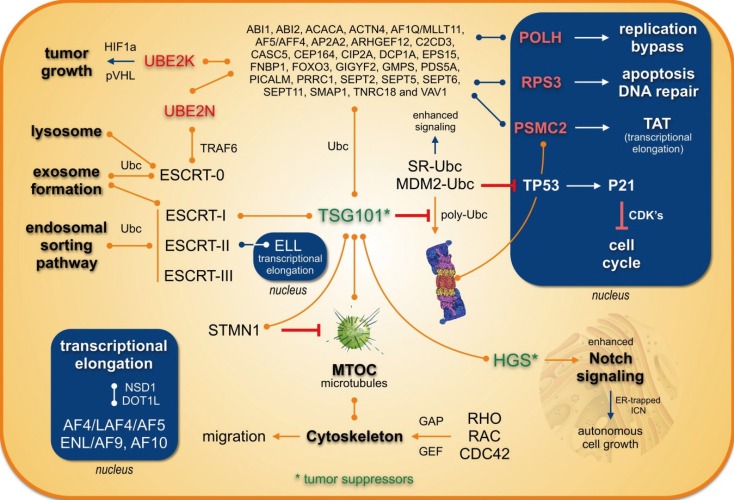
When analyzing the protein interaction network of all these proteins (BioGRID [thebiogrid.org] and STRING [string.embl.de] databases), another interesting observation can be made: a large number of MLL fusion partner proteins are linked to two particular protein networks (see Supplemental Data Fig. S1A-C). This is due to the fact that all of these proteins become mono- and/or poly-ubiquitinated which allow them to bind to certain proteins that are specialized to recognize this covalent protein modification. This seems to be common feature of many MLL fusion proteins and allow them to connect to the 'UBC-TSG101-HGS network'. Based on the data deposited in the BioGRID and the STRING database, they are also linked to a second protein network (UBC-PCNA-UBE2N-UBE2K-POLH-RPS3-PSMC2).
The following fusion partner proteins do show this particular feature: ABI1, ABI2, ACACA, ACTN4, AF1Q/MLLT11, AF5/AFF4, AP2A2, ARHGEF12, C2CD3, CASC5, CEP164, CIP2A, DCP1A, EPS15, FNBP1, FOXO3, GIGYF2, GMPS, PDS5A, PICALM, PRRC1, SEPT2, SEPT5, SEPT6, SEPT11, SMAP1, TNRC18, and VAV1 (n=28; see Fig. 6). They represent more than one third of all yet known MLL fusion partners.
THE UBC-TSG101-HGS NETWORK
Tumor susceptibility gene 101 (TSG101) represents an inactive Ubiquitin-conjugating enzyme that is specialized to recognize and bind via its UEV domain mono-ubiquinated proteins. When TSG101 binds to its 'target proteome', it prevents further poly-ubiquitination, and subsequently, the degradation of these target proteins.
In adddition, TSG101 binds to the microtubuli organizing center (MTOC) and is influencing microtubule and cytoskeleton formation. In particular, TSG101 has been first discovered as Stathmin-(STMN1/OP18)-interacting protein [74]. Stathmin is an important protein because it tightly regulates microtubule dynamics. Non-phosporylated Stathmin binds simultaneousy to two α/β-tubulin heterodimers and causes the bending of α/β-tubulin protofilaments, which in turn leads to a rapid disassembly of microtubuli. Phosphorylation of Stathmin at several serine residues (e.g. Ser-25 by RAC1) renders Stathmin inactive and allows microtubule or spindle formation. In addition, efficient microtubule formation requires CRMP2 that binds to single α/β-tubulin heterodimers and causes the polymerization of α/β-tubulin protofilaments. Interestingly, CRMP2 becomes inactivated by active GSK3β (see above). The kinase RAC1-activated by a complex composed of RhoG, ELMO2, and ILK-causes simultaneous phosphorylation of Stathmin and GSK3β in order to inactivate GSK3β. This in turn allows efficient microtubuli formation (phosphorylated Stathmin, non-phosphorylated CRMP2) [75]. After cytokinesis, Stathmin must be immediately dephosphorylated in order to allow the daughter cells to go into cell cycle arrest. Overexpression of Stathmin or the presence of a certain Stathmin mutation (Q18E) was found in many tumor cells and confer resistance against many chemo-therapeutics. Mutations in the STMN1 gene have a very strong oncogenic effect because they cause an uncontrolled cell proliferation [767778]. Moreover, hyperactive Stathmin destabilizes spindle microtubules, causing mitotic aberrancies, polyploidization, or chromosome losses.
The main function of TSG101 and HGS, however, is linked to the ESCRT System [79]. TSG101 mediates the association between the ESCRT-0 and ESCRT-I complex, and is usually involved in the sorting of mono-ubiquitinated proteins of the early endosomal pathway (vacuolar sorting). The ESCRT system is composed of three main complexes: ESCRT-I (Vps23=TSG101, Vps28, and Vps 37), ESCRT-II (Vps22, 2x Vps25, and Vps36), and ESCRT-III (Vps2, Vps20, Vps24, and Vps32). This system is also used by several viruses, like HIV, Hepatitis B, or measles virus, for their budding process via multivesicular bodies (MVB). The latter system is also important for exosome formation and secretion. Exosomes seem to have a great impact for cancer cells and are involved in manipulating/reprogramming of their surrounding, e.g. manipulating immune cells and to create a niche for tumor stem cells (for review see [8081]). The list of known proteins that are either found inside of exosomes or localized in the membrane of exosomes is huge, still growing, and comprises so far about 150 proteins (for review see [82]). Not to mention the role of certain microRNAs that are selectively secreted via exosomes to manipulate the niche of cancer stem cells (e.g. reviewed in [83]).
Apart from its function in vacuolar sorting, TSG101 is known to stabilize nuclear proteins, like MDM2 and steroid receptors. They all bind as mono-ubiquitinated proteins to the N-terminal UEV-domain of TSG101, thereby preventing their multi-ubiquitination and degradation via the proteasomal pathway. In case of MDM2, this leads to an increased degradation of p53 which causes a decrease of p21 and leads to an increased cell proliferation. Similarly, liganded nuclear receptors display extended transcriptional activities. TSG101 is overexpressed in many tumor cells to exert presumably this "non-ESCRT" function.
It is interesting to note that ELL, known as MLL fusion partner and important for transcriptional elongation, has first been purified as holocomplex together with Vps22, Vps25, and Vps36 which resembles the ESCRT-II complex [8485]. This links ESCRT proteins directly to nuclear processes like transcriptional elongation, and thus, causes effects known to be exerted by major MLL fusion partners (e.g. AF4, ENL, etc).
Another interesting aspect comes again from TSG101, HGS (=Vsp27), or Vps25 deficient cells. These cells display a sustained signaling activity from activated Notch receptors, because activated Notch (intracellular Notch [ICN]) becomes trapped in early endosomes. This leads to enhanced growth properties and changes in surrounding cells: they also start to proliferate and loose their cell polarity, a typical sign for a malignant conversion. Notch signaling is important for the development of normal cells as well as leukemic stem cells (for review see [86]).
In conclusion, many of the known MLL fusion partner genes express proteins that are linked to quite important pathways. In particular, the ESCRT system may provide some important clues for tumor cells. On one hand is steers vacuolar sorting of proteins, influence exosome formation, signaling processes, cell cycle, cytoskeleton dynamics, mitotic spindle system, and moreover, ESCRT-II/ELL has a direct function on transcriptional elongation (summarized in Fig. 6).
THE UBC-PCNA-UBE2N-UBE2K-POLH-RPS3-PSMC2 NETWORK
The second network seems to be less attractive at first sight in terms of cancer development. However, single proteins from that network are also quite interesting (marked by red letters in Fig. 6).
UBE2N is critical for correct B-cell development and macrophage activation and interacts with TRAF6. TRAF6 mediates the synthesis of lysine-63 linked polyubiquitin chains, which are not connected to the proteasomal degradation but connected to endocytic trafficking, inflammation, and DNA repair. Lysine-63 linked Ubiquitin chains are bound to the ESCRT-0 complex (Vps27=HGS and HSE1=STAM1/2) which is essentially involved in MVB formation. MVB are the precursor of secreted exosomes, or alternatively, for lysosome formation. Indeed, ESCRT-0 is the major pathway for the degradation of either misfolded or damaged proteins from the Golgi system. Decision between both pathways is made upon the presence of high amounts of cholesterol, ceramides, and flotillin (causes exosome production), while a low amount of cholesterol and the presence of lysobisphosphoric acid cause lysosome formation.
Polymerase eta (POLH) and the ribosomal protein RPS3 are both involved in DNA repair processes; POLH is involved in the repair of thymidin dimers, while RPS3 is involved in DNA base excision repair, induction of apoptosis, migration of cells and is again found as cargo in exosomes. Of interest, POLH is categorized in the "RAD6 epistasis group" which resembles the so-called "damage bypass group". Proteins of this RAD6 epistasis group lead to lysine-63-ubiquitination of PCNA (on its lysine-164 residue). This allows replication in the presence of DNA damage or thymidin dimers, because POLH incorporates correctly two A-nucleotides during replication even when a thymidin dimer is present at the opposite strand.
RPS3 is a ribosomal protein and makes connections to a large variety of other proteins in cells. The list of interaction partners includes NFkB1, PRMT1, TP53/MDM2, and the RB protein. RPS3 is also a target of several signal transduction pathways (AKT, ERK, and PKCδ) that link RPS3 to several functions: both PKCδ (S-6 and T-221) and ERK phosphorylation (T42 residue) are associated with DNA repair processes, while AKT phosphorylation (T70 residue) leads to cell death. RPS3 overexpression leads to a degradation of PCNA and LaminA/C which is typically observed by triggering the apoptic pathway (for review see [87]).
PSMC2 is a regulatory subunit (AAA-ATPases) of the 26S proteasome and influences the differentiation and apoptotic behavior of cells. More interestingly, PSMC2 binds the TAT protein and influences again the process of transcriptional elongation.
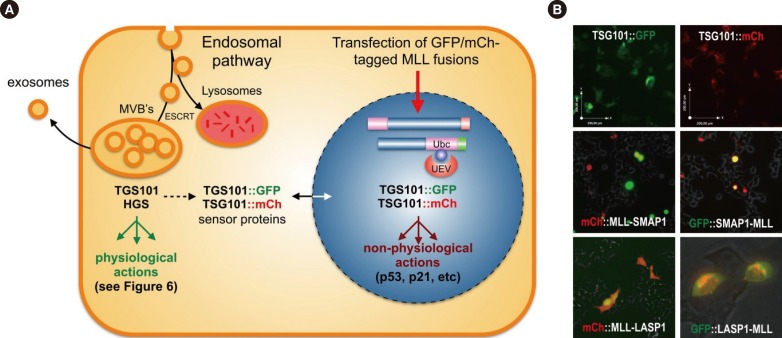
In conclusion, many of the yet known MLL recombination partner proteins-most of them localizing to the cytosol-are now enabled to enter the cell nucleus when fused to the MLL protein. This may cause a delocalization of their cognate binding proteins as well. For TSG101, this is an already known feature, because it is mainly localized in the cytosol, but can enter the nucleus upon binding to certain binding partners. In the artificial situation of a chromosomal translocation, a nuclear re-localization of TSG101 may lead to the deregulation of the cytosolic pathways (migration, apoptotic behavior, cell growth, signaling pathways), while its nuclear function becomes overt. As summarized in Fig. 7, two color-tagged TSG101 reporter proteins (either red or green) are shuttling into the nucleus when MLL fusion proteins were co-expressed (MLL-SMAP1, MLL-LASP1, SMAP1-MLL). In one case, LASP1-MLL, we saw retention in the Golgi aparatus. However, in all investigated cases, TSG101 becomes depleted in the cytosol. This underlines our notion that the delocalization of TSG101-which controls the abundance of many other proteins-could be a novel mechanism to explain the oncogenic effects exerted by MLL fusion proteins.
CURRENT DIRECTIONS
So far, most scientific activities are concentrating on direct MLL fusions and their associated proteins which display per se interesting target structures. Exemplarily, inhibition of the interacting DOT1L histone methyltransferase or the MLL/MEN1 protein-protein interaction at the N-terminus of MLL has been used to design very specific drugs. No doubt these new drugs (EPZ5676 and MI-503/MI-463) [8889], which are in clinical trials, will have an impact on the treatment of MLL-r leukemia patients. However, these drugs will also harm normal cells, although this will presumably not be easily visible in short-term treatments or short-term animal experiments. However, it can be predicted that stem cells, especially hematopoietic stem cells, will be impaired by the treatment. The reason for this is outlined above, because wild type MLL functions are necessary for their maintenance. In the context of treatment strategies that act directly on MLL or MLL-associated proteins, it is interesting to mention that haplo-insufficiency of MLL is associated with the recently described Wiedemann Steiner syndrome [90]. These rare patients express MLL only from a single allele, because of a premature stop codon at the second allele and the destruction of the mutant transcripts via an RNA decay mechanism. However, producing MLL only at a 50% dosage already results in a phenotype termed hypertrichosis cubiti (hairy elbows), intellectual disability, a short stature, small kidneys, and a distinctive facial appearence. This should be kept in mind when treating patients with inhibitors that target the MLL complex.
Therefore, alternative or additive treatment options should be taken into account. Recently, we have shown that class I HDAC inhibition is able to reverse the oncogenic activity mediated by MLL-AF4. This is due to the fact that endogenous or plasmid-transfected MLL becomes converted from its CYP33 bound "inactive state" into its "active state" upon HDACi treatment. The HDACi-enhanced MLL complexes caused the displacement of MLL-AF4 at target gene promoters and completely reversed the enhanced transcription on MLL target genes [91]. Activating endogenous MLL by HDACi may also compensate for the expected negative side effects when using MLL/MEN1 inhibitors. Therefore, it should be considered to combine HDACi treatment with those experimental drugs in order to avoid severe side effects.
OUTLOOK
This review tried to provide a rational explanation why so many different MLL fusion partners all contribute to the onset of acute leukemia and display very similar clinical courses. In first instance, any of the yet known rearrangements of the MLL gene leads to a destruction of the intrinsic "CYP33-switch" of the MLL protein. The physical separation of MBD- and PHD-domains causes a loss of this important control unit. Consequently, the two resulting fusion proteins are acting in a dominant-positive manner over their wild type counterparts.
Secondly, the data presented here also point to a second mechanism, because a large portion of the yet known MLL fusion partner proteins share common pathways. On the basis of this functional compilation, a large portion of known MLL fusion partners may be involved in cytoskeleton/microtubuli signaling pathways. A third mechanism derives from the relocalization of proteins or domains thereof to the nucleus. In addition, many of those proteins are targets of ubiquitination pathways which may also influence or compromize the TSG101 protein. TSG101 exhibits the specific UEV-domain which allows binding to mono-Ubc proteins, thereby blocking poly-ubiquination. This in turn enhances the stability of bound proteins and changes their half-life. On the basis of preliminary data with color-tagged MLL fusions (mCh::MLL-SMAP1, mCh::MLL-LASP1, GFP-SMAP1-MLL, and GFP::LASP1-MLL) and TSG101 reporter cell lines (TSG101::GFP or TSG101::mCh), we propose the hypothesis that MLL fusion proteins lead to a translocation of TSG101 into the nucleus or another organelle (see Fig. 7A and B). Nuclear TSG101 has a protective function for these MLL fusions or other transcription factors (see above), thereby impairing p53 and p21. If this happens, many other nuclear (enhanced half-life) and cytosolic proteins (reduced half-life) become deregulated in their abundance. One example is the already mentioned nuclear receptors or members of the AFF family, like AF4/AFF1, LAF4/AFF2, and AF5/AFF4, which are known to become ubiquitinated. We already know that a stabilization of these proteins causes ectopic transcriptional activation due to enhanced transcriptional elongation [313392]. Enhanced transcriptional elongation and ectopic epigenetic histone signatures are two important mechanisms that accompany the process of malignant transformation.
A final scenario concerns exosome formation. It becomes more and more evident that exosomes play an important role in many different cancer types. They are used to manipulate surrounding (immune) cells or for niche formation (for review see [8081]). Oncogenic stress, which may occur owing to expression of MLL fusion proteins, is a known factor to increase exosome formation, quite similar to what happens during viral or bacterial infections. These stress-mediated reactions depend on intracellular Ca2+ signaling, P2X7 receptor signaling via ATP, and the ceramide biosynthesis pathway. Interestingly, SNP mutations in P2X7 are already known to be linked to the onset of CLL, but not for ALL or MLL-r leukemia. Whether the presence of MLL fusion proteins is directly or indirectly impacting exosome secretion is currently unknown. However, it will be worth investigating this new direction, because of TSG101 and HGS have this interesting relationship with the above described fusion partner proteins as well as with exosome production.
In conclusion, we understand functionally only a handful of the many fusion partner proteins; however, even this little knowledge is exciting and holds many promises for the treatment of this particular group of leukemia patients. Right now, many scientists are focusing on approaches that follow the experimental results obtained with MLL-ENL and MLL-AF9 and their role in causing AML in model systems. However, we have many other MLL gene fusions that need definitively more experimental attention. The most prominent MLL-r leukemia derives from t(4;11) translocations, and here, more questions than answers are currently present. The debate on the necessity of the reciprocal AF4-MLL fusion protein for t(4;11) leukemia is still dividing the scientific community [9599394], but more and more data becomes available that substantiate the importance of reciprocal MLL fusions [959697]. Even when genome editing techniques were applied to create t(9;11) or t(4;11) chromosomal translocations as a single hit, both MLL fusion proteins were required for the long-term survival of these transformed cells [98]. Therefore, we should extend our targeting concepts to reciprocal MLL fusions. First approaches have already been published that are targeting AF4-MLL-but not MLL [99]. Also, the recently published SET-inhibitors are quite interesting [100], but this will again interfere with wild type MLL. Therefore, we need to follow up already existing strategies, but will need to find new targets or pathways in order to treat MLL-r leukemia.
 XML Download
XML Download