

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pancreatitis is characterized by inflammation of the pancreas that progresses from acute (sudden onset; duration less than 6 months) to rec urrent acute (more than one episode of acute pancreatitis) to chronic (duration more than 6 months). Repeated episodes of acute pancreatitis can progressively lead to irreversible exocrine and endocrine pancreatic insufficiency. Gallstones and alcohol abuse are the two most common causes of acute pancreatitis, and alcohol abuse is the most frequent cause of chronic pancreatitis. However, the etiology of 10-30% of acute or chronic pancreatitis cases cannot be readily identified and these cases are classified as idiopathic pancreatitis [1].
In 1996, the cationic trypsinogen gene PRSS1 (OMIM #276000; NM_002769.4) was identified as a cause of hereditary pancreatitis (HP) and some cases of idiopathic chronic pancreatitis (ICP) [23]. HP is a rare autosomal dominant disorder characterized by recurrent attacks of pancreatitis, with approximately 80% penetrance by 20 yr of age [4]. PRSS1 encodes cationic trypsinogen, the most abundant isoform of trypsinogen in human pancreatic juice. Pathogenic PRSS1 variants alter the activation and degradation of cationic trypsinogen by chymotrypsin C (CTRC; OMIM 601405; NM_007272.2) [5]. The p.R122H and p.N29I of PRSS1 have been identified as the most common pathogenic variants that exhibit high penetrance and follow the autosomal dominant pattern of pancreatitis [6]. Gain-of-function pathogenic PRSS1 variants, including pathogenic missense mutations and pathogenic copy number variations (CNV) such as gene duplication or triplication, have been identified in ICP patients [7].
In addition, pathogenic variants of the cystic fibrosis transmembrane conductance regulator (CFTR; OMIM 602421; NM_000492.3) [89], serine protease inhibitor Kazal type 1 (SPINK1; OMIM 167790; NM_003122.4) [1011], and CTRC genes have been described in both ICP and alcoholic CP [12]. Therefore, mutational analysis may reveal a genetic basis for idiopathic pancreatitis in a significant proportion of patients. Indeed, genetic testing may play a critical role in the evaluation and management of pancreatitis.
The four above-mentioned genes have been the most extensively studied genes to date that promote or cause pancreatitis. However, few reports [1314] have studied all four genes simultaneously in idiopathic pancreatitis. In addition, pathogenic variants of CTRC and CNVs of PRSS1 and SPINK1 have not been evaluated in Korean patients with pancreatitis. In this study, we identify the spectrum and frequency of pathogenic variants of PRSS1, SPINK1, CFTR, and CTRC and CNVs of PRSS1 and SPINK1, and determine their association with the clinical course of idiopathic pancreatitis in Korean patients.
Go to : 
METHODS
1. Patients
The study population consisted of 116 Korean subjects (65 males, 51 females; mean age, 30.4 yr, range, 1-88 yr) diagnosed with ICP (n=59), idiopathic recurrent acute pancreatitis (IRAP, n=14), or idiopathic acute pancreatitis (IAP, n=43). Only patients with pancreatitis confirmed by their referring physicians from July 2008 to December 2015 were included in this study. Written informed consent was obtained from each patient, and this study was approved by the Institutional Review Board of Gangnam Severance hospital, Seoul, Korea. Clinical information was obtained for 84 of the patients via a retrospective review of medical records. Imaging examination, including endoscopic retrograde cholangiopancreatography (ERCP), computed tomography (CT), magnetic resonance imaging (MRI), ultrasonography, and magnetic resonance cholangiopancreatography (MRCP), was performed to detect changes in the pancreatic duct (stenosis or dilation). The majority of patients (58 of 84, 69%) expressed abdominal pain, four patients exhibited weight loss, two patients had abdominal discomfort, and one patient had portal hypertension as their chief complaint on the first doctors' visit. Interestingly, 20 patients were diagnosed with chronic pancreatitis during medical examination or during evaluation for co-morbidity. During the evaluation for pancreatitis, eight patients were diagnosed with pancreatic cancer and one was diagnosed with intraductal papillary mucinous neoplasm. The majority of pancreatic cancer patients had CP (n=6). Imaging examination detected pancreatic duct changes (stenosis or dilation) in 26 patients, and pancreatic duct stones, pancreatic calcification, and pseudocysts in nine patients each. In addition, annular pancreas, pancreatic divisum, and anomalous union of pancreaticobiliary duct (AUPBD) were detected in one patient each (Table 1).
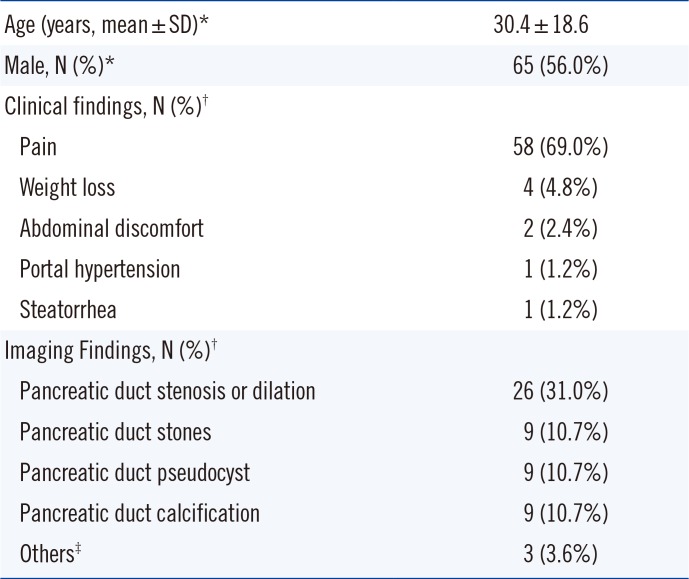
Table 1
characteristics of patients with idiopathic pancreatitis
![]()
2. DNA isolation
Genomic DNA was extracted from EDTA-treated whole blood samples by using a QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany), on a QIAcube automatic nucleic acid extraction instrument (Qiagen), according to the manufacturer's instructions.
3. Analyses of PRSS1, SPINK1, CFTR, and CTRC pathogenic variants
Genomic DNA was amplified by PCR and sequenced by the Sanger method. Sequence analysis of PRSS1 (exon 2, 3, and 5), SPINK1 (exon 3), CFTR (exon 3 and 7), and CTRC (exon 25 for Q1352H) was performed on an ABI 3500xL system (Applied Biosystems, Foster City, CA, USA).
4. Detection of copy number variations
Multiplex ligation-dependent probe amplification (MLPA) was performed to assess PRSS1 and SPINK1 CNVs, using an MLPA kit (SALSA MLPA KIT P242 Pancreatitis, MRC Holland, Amsterdam, The Netherlands) according to the manufacturer's instructions. MLPA fragment analysis data were generated on an ABI 3500xL system (Applied Biosystems) and analyzed by using GeneMarker (SoftGenetics, State College, PA, USA).
5. Statistical analysis
The Student's t-test (unpaired) was performed for comparing quantitative data, and χ2 test or Fisher's exact test was performed for analyzing qualitative data. A P value less than 0.05 was considered statistically significant. SPSS version 20 (IBM, Armonk, NY, USA) was used for statistical analysis. We examined whether the clinical course of pathogenic variant-positive and pathogenic variant-negative patients was significantly different. We reviewed the clinical course, including the clinical features and imaging findings.
Go to :
RESULTS
1. Genetic analysis
All 26 idiopathic pancreatitis cases (22.4%) carried at least one causative or contributory genotype (Table 2). We found three types of pathogenic PRSS1 variants in 11 patients, including p.N29I (n=1), p.R122H (n=1), and p.G208A (n=9). Among patients with PRSS1 p.G208A, SPINK1 c.194+2T>C (n=2), SPINK1 p.N34S (n=1), and CFTR p.Q1352H (n=4) were also detected. We found three types of heterozygous pathogenic SPINK1 variants in 16 patients, including c.194+2T>C (n=12), p.N34S (n=3), and c.194+1G>A (n=1). Two patients with SPINK1 c.194+2T>C and p.N34S also had CFTR p.Q1352H. A single heterozygous CFTR p.Q1352H pathogenic variant was detected in eight patients. One patient carried a heterozygous CTRC p.P249L (rs142560329) variant, which was previously established to be a high-risk CTRC variant by functional analysis [15]. A rare CTRC p.R246C variant was also detected in one patient. PRSS1 and SPINK1 gene copy numbers were normal in all patients tested (n=93).
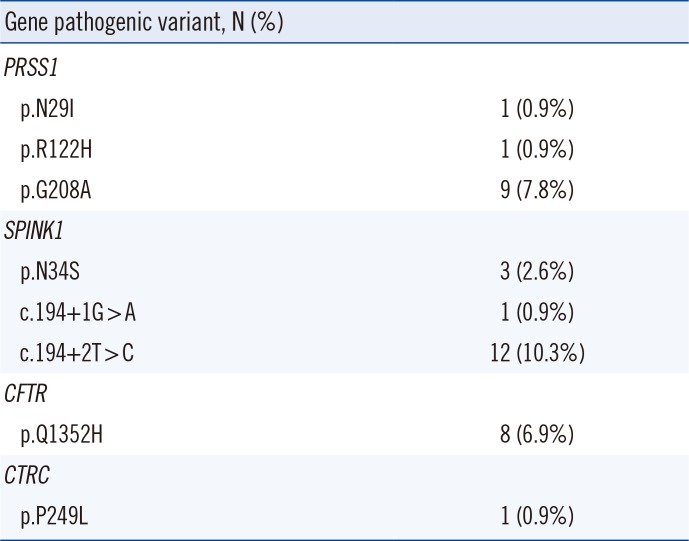
Table 2
Pathogenic variants of PRSS1, SPINK1, CFTR, and CTRC in patients with idiopathic pancreatitis
![]()
2. Association of pathogenic variants with clinical course
We associated genotype data with clinical data from 84 patients (Table 3). The c.194+2T>C pathogenic variant in SPINK was the most common variant, followed by the p.G208A pathogenic variant in PRSS1 and the p.Q1352H pathogenic variant in CFTR. However, only pathogenic variants of PRSS1 and SPINK1 were associated with clinical courses. Weight loss occurred more frequently in patients with the p.G208A pathogenic variant (2.6% vs 25.0%; P=0.044), while pancreatic duct stones occurred more frequently in patients with the c.194+2T>C pathogenic variant (6.8% vs 40.0%; P=0.01). Although the difference was not statistically significant, pancreatic stenosis occurred more frequently in patients carrying the CFTR p.Q1352H pathogenic variant (28.2% vs 66.7%). Pancreatic duct stones were more common in patients with the PRSS1 p.G208A pathogenic variant (7.9% vs 37.5%). We did not find an association between any of the pathogenic gene variants (PRSS1, SPINK1, CFTR, and CTRC) and an earlier age of symptom onset. Additionally, the dosage of pathogenic variants was not associated with the clinical course. Among eight pancreatic cancer patients, two had pathogenic variants (SPINK c.194+1G>A and CFTR p.Q1352H, respectively). However, the clinical course of pathogenic variant-positive patients was not different from that of pathogenic variant-negative patients, except for those diagnosed with cancer at a younger age.
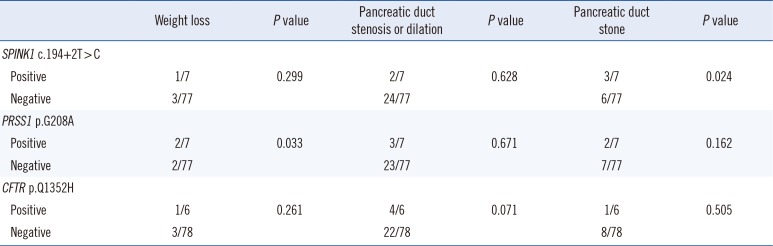
Table 3
Association of pathogenic variants with clinicopathological data from 84 patients
![]()
Go to :
DISCUSSION
Schnur et al [16] reported that PRSS1 p.G208A pathogenic variant was rare and predominant in Asia, and caused a moderate secretion defect [16]. Lee et al [17] reported that it was the most common pathogenic variant detected in Korean children with HP. The p.G208A pathogenic variant is considered a milder pathogenic variant than the PRSS1 'classic pathogenic variant,' p.R122H, because of its association with alcoholic CP and the co-existence of the CTRC p.R29Q and the SPINK1 p.N34S in some patients [18]. Indeed, some patients carrying the p.G208A pathogenic variant also harbored pathogenic variants of SPINK1 or CFTR. In a previous study, a patient with pancreatitis and carrying the p.G208A pathogenic variant also harbored p.F508del and p.Q1352H pathogenic variants of CFTR [19]. Importantly, our results show that the PRSS1 p.G208A pathogenic variant is associated with idiopathic pancreatitis in Korean children and adults. The patients carrying the p.G208A pathogenic variant presented weight loss more frequently compared with patients lacking this pathogenic variant. Weight loss can occur owing to loss of pancreatic function or decreased food consumption due to postprandial pain.
Patients with pathogenic variants of PRSS1 are at a greater risk of developing pancreatic cancer, owing to the prolonged history of CP [20]. While we were unable to identify any association of pathogenic variants with the development of pancreatic cancer, it should be noted that the two cancer patients with pathogenic variants were younger than the other patients without variants (41.5 yr vs 59.8 yr, P=0.033). Therefore, we believe that the pathogenic variants responsible for CP might play a role in the early development of pancreatic cancer.
SPINK1 is a potent anti-protease that functions as a major inactivator of intrapancreatic trypsin. SPINK1 pathogenic variants act as a risk modifier in recurrent acute pancreatitis, thereby lowering the threshold for developing chronic pancreatitis induced by other genetic or environmental factors [21]. In agreement with previous studies [2223], the frequency of the SPINK1 c.194+2T>C pathogenic variant was much higher than the frequency of the p.N34S heterozygous pathogenic variant that occurs commonly in western countries [24]. The c.194+ 2T>C pathogenic variant resulted in patients manifesting a more complicated disease pathology than patients lacking this pathogenic variant. A novel pathogenic splicing variation, c.194+1G>A, was found in a 41 yr old man diagnosed with pancreatic duct stones and CP. This variant affects an intron splice donor site by altering the highly conserved GT dinucleotide. Although the actual effect of the SPINK1 variant c.194+1G>A was not investigated, this novel splicing variation was classified as 'likely pathogenic' by the ACMG standards and guidelines for the interpretation of sequence variants [25].
Masson et al reported gain-of-function duplication and triplication of an approximately 605-kb segment containing PRSS1 and PRSS2 on chromosome 7q34 in French patients with hereditary or idiopathic CP [726]. In addition, they identified two large genomic deletions in SPINK1 that caused chronic pancreatitis [2728]. Recently, CNVs of PRSS1 were also reported in Chinese ICP patients, wherein four patients carried only one copy and one carried five [13]. In contrast, no CNVs were identified in this study.
Pancreatic insufficiency in patients with cystic fibrosis is due to pathogenic variants of the CFTR gene, such as p.F508del, p.R117H, and p.N1303K, which result in a defective chloride channel and subsequent pancreatic duct obstruction. Pathogenic variants of CFTR are associated with chronic pancreatitis without any evidence of cystic fibrosis [89]. A previous study [29] reported that the pathogenic CFTR p.Q1352H variant in the Korean population was significantly higher in patients with bronchiectasis and chronic pancreatitis than in the control group. In addition, the authors reported that the pathogenic p.Q1352H variant decreased mature CFTR protein expression by 73% and chloride current activity by 71%. Therefore, they suggested that the p.Q1352H pathogenic variant was strongly associated with respiratory and pancreatic disease among Koreans. CFTR p.Q1352H variant observed in eight patients. The allele frequency in control groups from Jang et al [30] was 1.2%, which is similar to publicly accessible data (0.49 to 2.4% from 1,000 genomes; 1.3% from ExAC Browser). The pathogenic p.Q1352H variant was significantly associated with ICP (P=0.039) in our study.
Rosendahl et al [12] analyzed CTRC gene variants in a German cohort of over 300 patients with idiopathic or hereditary CP and revealed that alterations in CTRC predispose patients to pancreatitis by decreasing the protective, trypsin-degrading activity of this enzyme. Pathogenic CTRC variants in exons 3 and 7 were also associated with CP in Europe and India [1231], although the spectrum of the pathogenic variants was different between the two populations. Interestingly, the spectrum and distribution of pathogenic CTRC variants were different within Asian subjects. Masamune et al [32] found five novel missense variants (8/506) in Japanese CP patients. However, only three variants, including p.R29Q, p.S239C, and p.R254W, were shown to be functionally deleterious [1533]. A cohort study with Chinese pediatric ICP patients [13] did not find any pathogenic variants of CTRC, whereas we identified one patient with the pathogenic p.P249L variant. Taken together, CTRC pathogenic variants are uncommon and do not play a significant role in idiopathic pancreatitis in the Korean population.
The current study had several limitations. First, we could not determine the natural history of IP complications based on these data and the limited subgroups. Additionally, we sequenced only targeted regions of the PRSS1, SPINK1, CFTR, and CTRC genes. However, we were able to identify the pathogenic variant spectrums of these genes in Korean IP patients, and we were able to associate pathogenic variants with clinicopathological data.
In conclusion, we report that PRSS1, SPINK1, and CFTR pathogenic variants were associated with IP, whereas pathogenic variants of CTRC were not associated with this disease. We did not detect CNVs for PRSS1 and SPINK1 in Korean patients with idiopathic pancreatitis. Patients with pathogenic variants of PRSS1 and SPINK1 presented a more severe clinical course, implying that genetic risk assessment may be useful for identifying individuals likely to develop severe CP, and develop targeted therapy for slowing or preventing disease progression. Larger prospective studies to evaluate the clinical impact of pathogenic variants are needed, along with comprehensive analyses by next generation sequencing to identify novel pancreatitis-associated genes.
Go to :
 XML Download
XML Download