

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
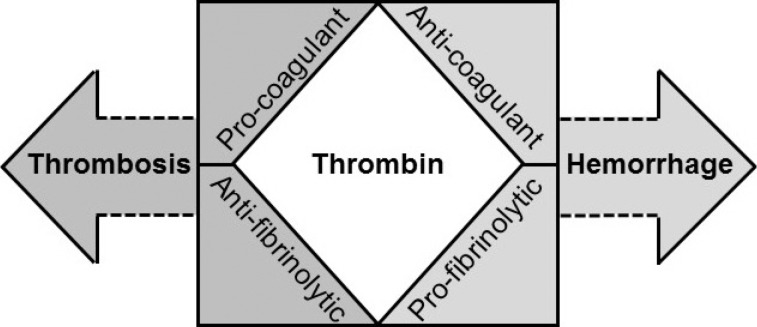
Normal hemostasis is a well-orchestrated process whereby adequate amounts of thrombin are generated at the site of vascular injury. This results in the formation of a firm stable clot involving tight coordination between pro- and anticoagulant pathways as well as pro- and antifibrinolytic systems to maintain circulatory flow without perturbation from the growing thrombus [1]. These physiological processes are pathologically altered to varying degrees in patients developing disseminated intravascular coagulation (DIC). In DIC, an excess of thrombin is generated and continued by inciting factors to overwhelm the homeostatically controlled hemostatic process (Fig. 1), which can then disseminate systemically [2]. This concept has been taken into consideration by the International Society of Thrombosis and Haemostasis (ISTH) Scientific and Standardization Committee, which defines DIC as "an acquired syndrome characterized by intravascular activation of coagulation with loss of localization arising from different causes. It can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction" [3]. This statement from collective experts in the field summarizes our understanding of the pathogenic concepts involved and shape our clinical considerations and management.
Go to : 
PATHOPHYSIOLOGICAL CONSIDERATIONS
As DIC is an intermediary mechanism of disease from conditions such as sepsis, trauma, obstetrical calamities, or cancer, there will be disease-specific pathogenic factors that influence both coagulation activation and its functional consequences [4]. A major challenge in treating patients with DIC is in identifying the predominant mechanism among the heterogeneous overlapping effects, which can also vary with time. Key themes for the clinician to consider (Fig. 2) include.
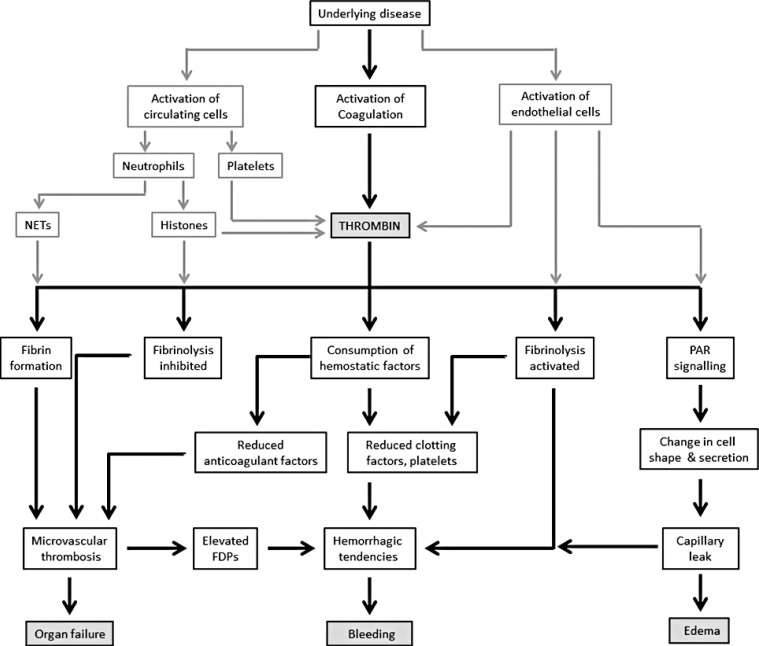 | Fig. 2Pathophysiological considerations in the clinical presentation of DIC. Systemic activation of coagulation together with cellular activation (endothelial cells, neutrophils, and platelets) leads to excessive thrombin generation and its functional consequences.Abbreviations: FDP, fibrin degradation products; NETs, neutrophil extracellular traps; PAR, protease-activated receptor.
|
1. The multifaceted role of thrombin generation in vivo
Understanding the diverse and opposing thrombin-orchestrated effects is important in judging and refining management approaches when the fine homeostatic balance of normal thrombin generation is lost in DIC. Thrombin is typically associated with procoagulant properties in cleaving fibrinogen into fibrin and also activating platelets, on which tenase (FIXa/VIIIa) and prothrombinase (FXa/Va) complexes assemble to propagate coagulation [5]. Conversely, thrombin also has anticoagulant properties through thrombomodulin (TM)-dependent protein C (PC) activation, and these mechanisms are well reviewed elsewhere [6]. Equally, thrombin is also pivotal in affecting opposing aspects of fibrinolysis, i.e., promoting through stimulating endothelial cell release of tissue plasminogen activator (tPA) [7] to generate plasmin, and conversely, inhibiting through plasminogen activator inhibitor 1 (PAI-1) [8] induction and thrombin-activatable fibrinolysis inhibitor (TAFI) activity [9]. Whilst the DIC process could consume these different pathway components proportionately, the triggering pathology can influence the rate of thrombin generation and direct its function [10]. One example of disease-specific tailoring of DIC is the significantly more intense coagulant response in early trauma. Together with the extensiveness of endothelial cell activation, the excessive surge in thrombin leads to overdrive of tPA-triggered fibrinolysis and indiscriminate fibrin(ogen)olysis, as observed in a baboon model of DIC following blockade of the PC anticoagulant pathway to enhance thrombin generation [11].
2. Concomitant cellular dysfunction, especially in the microcirculation
Cell surfaces and site-specific vascular endothelial features control the processes consequent to thrombin generation [12]. The microvasculature is particularly affected in DIC because of its higher endothelial-cell surface to blood volume ratios [13]. Receptors of the PC pathway, i.e., the endothelial PC receptor (EPCR) and TM, are among the most relevant as both have direct pleiotropic effects on coagulation and inflammation. Activated protein C (APC) when bound to EPCR can directly cleave and activate protease-activated receptor 1 (PAR1) to generate anti-inflammatory, antiapoptotic, and endothelial-barrier-protective effects [14]. These are directly opposite to those generated by thrombin on PAR1 and exemplify the yin-yang balance between thrombin and APC in harmonizing the coagulation and inflammatory processes at the blood-endothelial interface [6]. The lectinlike domain of TM has been implicated in reducing the proinflammatory effects of high-mobility group box protein 1 (HMGB1) released from the nucleus [15]. Consequently, loss of EPCR and TM through receptor shedding or endothelial down-regulation in microvessels with low constitutive expression could shift the thrombin–PC axis toward site-specific thrombosis and barrier destabilization. This has been reported in cerebral malaria with DIC [16], especially as Plasmodium falciparum-infected erythrocytes bind EPCR to reduce APC generation [1718].
3. The overlapping contribution of innate immune activation and inflammation
Significant cross-talk between these components with the coagulant response has been increasingly characterized as immunothrombosis [1920]. With cell damage and death common to all the causes of DIC, there is triggering of damage-associated molecular patterns (DAMPs) leading to promotion of thrombin generation [21]. Cross-activation of pathways such as the complement system [22] results in reciprocal escalation causing further cell damage and an increased release of nuclear breakdown products, such as histone-DNA complexes (nucleosomes) and double-stranded DNA [23]. Elevated circulating histone levels occur in patients with DIC [24], and mechanistically, they bind phospholipid surfaces to damage cell membranes [2526]. Functional consequences include platelet aggregation and thrombocytopenia [27], thrombi that are more resistant to lysis [28], and vascular leakage as well as a release of proinflammatory cytokines and extracellular traps from leucocytes, especially neutrophils [25]. Referred to as neutrophil extracellular traps (NETs), these are prothrombotic [29] by providing a scaffold for assembling clot components including tissue factor, degrading tissue factor pathway inhibitor (TFPI), and histone-dependent Factor VII-activating protease activity [30] as well as a platelet polyphosphate release [31]. In addition to suppressing TM-dependent PC activation [32], histone 4 can induce prothrombin autoactivation in vitro [33]. Such a mechanism would generate thrombin independently of activation of the coagulation cascade if present in vivo. The infusion of histones in experimental models leads to DIC with the spectrum of hemorrhage and microvascular thrombosis [2425]. Blocking histones reduces thrombin generation [25] and ameliorates cardiac dysfunction and acute lung injury in murine models of sepsis [34] and trauma [25], respectively.
Go to :
THE CLINICAL PRESENTATIONS OF DIC
In general, physicians tend to consider the possibility of DIC only when there is extensive and uncontrollable bleeding from multiple sites. As mentioned earlier, hybrid pathology with increasing bruising or bleeding and ischemic changes secondary to microvascular thrombosis is characteristic of DIC. Bleeding can be secondary to various reasons including thrombocytopenia, platelet dysfunction, consumption of clotting factors, increased vascular permeability, excessive fibrinolysis, and interference of fibrin degradation products (FDPs) with clot firmness. Thrombosis can be secondary to reduced levels of anticoagulant factors, platelet activation, NETs, histone-induced cell damage, and procoagulant effects. Clinically, this may be subtle, especially with insidious microcirculatory obstruction leading to organ dysfunction.
Go to :
THE LABORATORY ASPECTS
1. General principles
DIC is a clinical-laboratory diagnosis, and laboratory changes need to be interpreted with knowledge of the patient's underlying disorder [35]. In a relevant clinical scenario, several laboratory parameters are analyzed together as part of a diagnostic algorithm that includes prothrombin time (PT), activated partial thromboplastin time (aPTT), the platelet count, fibrinogen level, and a marker of fibrin degradation, e.g., D-dimer or the soluble fibrin monomer (SFM) [35]. None of these markers are taken in isolation, and a combination of results at different time points is particularly helpful in determining the presence of DIC (Table 1). It must also be emphasized that certain profiles may be more indicative of certain pathological mechanisms, e.g., hyperfibrinolysis is typically characterized by very low fibrinogen levels with very high concentrations of FDPs.

Table 1
Disseminated intravascular coagulation (DIC) International Society of Thrombosis and Haemostasis (ISTH) scoring criteria
![]()
2. Composite scoring systems
The ISTH scoring system of routine global coagulation tests provides a framework for diagnosing DIC that also standardizes criteria for clinical studies. This is now well validated in different settings, and a cumulative score of 5 or more from increased PT, reduced platelet counts and fibrinogen levels, together with elevated FDPs forming the basis of our approach [36]. There are country-specific refinements [37], which similarly confirm the independent prediction of mortality by the ISTH DIC score and its added prognostic value to the Acute Physiology and Chronic Health Evaluation (APACHE) II system [438]. Specific caveats for the individual parameters are discussed in the sections below, and our practice relies on a high index of suspicion for increasing abnormalities, with particular emphasis on serial monitoring. Conceptually, this is encapsulated as "nonovert DIC" by the ISTH, where subtle hemostatic dysfunction could provide forewarning of an overt DIC process [3]. Indeed, several studies have shown how scoring for abnormal trends in PT, platelets, and/or D-dimer has identified patients deteriorating clinically with new organ failure [394041]. Clinicians and laboratory staff who are caring for critically ill patients with disorders that can potentially trigger DIC (e.g., sepsis, trauma, malignancy, and certain obstetrical conditions) should be vigilant to this aspect of management. The presence of "normal" values for PT, platelets, and/or D-dimers in such patients upon admission should not mean the end of monitoring of these parameters. Awareness of the possibility of DIC should trigger serial monitoring of these parameters, and other measurements may be requested to confirm nonovert DIC (see the section below). This strategy provides a safeguard for the early recognition of DIC in such patients, with a major impact on management and prognosis.
3. Specific test considerations
There are certain caveats in interpreting results from the recommended tests:
1) PT and aPTT
In general, physicians only consider a diagnosis of DIC when the PT and aPTT are considerably prolonged. Since aPTT can be shortened owing to increased Factor VIII levels from the acute phase response, aPTT prolongation may lag behind the clinical manifestations [42]. There is also the potential influence of heparin and the considerable variation in sensitivity of aPTT reagents [43]. As such, the ISTH DIC Subcommittee recommends PT as the coagulation test of choice [3644], and in laboratories where PT in seconds is not reported, the international normalized ratio (INR) could provide reliable information [45]. Although the different sensitivities of thromboplastin reagents to coagulation defects induced by DIC and vitamin K antagonists need to be considered, the use of human recombinant thromboplastins generally negates these differences. Even with the greater variability of tissue-derived thromboplastins, the conversion into a limited point range from 0 to 2 within the ISTH scoring system minimizes any variation among thromboplastins [45]. It is also important to recognize that prolonged PT in DIC may also be due to other etiologies such as vitamin K deficiency or liver disease, which may not improve with treatment of DIC.
2) Platelet count
Thrombocytopenia is the commonest laboratory diagnostic feature of DIC [42] but may also be due to drugs, hemodilution, marrow failure, splenomegaly, or infections [46]. In DIC, platelet activation can be a source of inaccuracy when using automated platelet counting [47]. Thrombocytopenia in DIC is often accompanied by abnormal platelet function with functional defects enhancing the bleeding risk and heightened platelet aggregation [48], with a release of procoagulant components able to promote thrombosis. The finding that the von Willebrand factor-cleaving protease ADAMTS-13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) can be reduced in patients with DIC [49] may be particularly relevant in the latter scenario and has been linked to organ dysfunction, especially renal impairment [50]. Furthermore, thrombocytopenia is but one snapshot of the vicious cycle of events that could be histone mediated in amplifying cell death and dysfunction and propagating thrombin generation. In intensive-care settings, high levels of circulating histones on admission are significantly associated with the development of subsequent moderate-to-severe thrombocytopenia [51]. Of interest is the likelihood of multicomponent interaction in vivo involving NETs as activated platelets can also stimulate NETosis [52]. This needs careful consideration ahead of transfusing platelets purely on the basis of the count, especially as there is now evidence that this could promote thrombosis and death in consumptive disorders [53].
3) Fibrinogen level
Rather than a low absolute value, e.g., less than 1 g/L, which has a low sensitivity (22%) and high specificity (100%) for DIC [54], we tend to give importance to a decreasing fibrinogen level as it is also an acute-phase reactant [44]. It is important that the laboratory use a Clauss fibrinogen assay with awareness that the presence of high FDPs can interfere with clot endpoints and artefactually lower fibrinogen estimates [55].
4) Fibrin-related markers
As prolonged PT and thrombocytopenia are also seen in liver disease, the finding of elevated levels of biomarkers of fibrin formation can help to diagnose DIC. In terms of which fibrin-related marker to use, SFM is theoretically a better early indicator of coagulation activation in reflecting the intravascular action of thrombin on fibrinogen [56]. This appears to hold true in intensive care settings: with SFM being more sensitive than FDPs or D-dimer in detecting early abnormalities in PT and platelet counts. However, the downstream markers of fibrin formation, i.e., FDP and D-dimer, are more statistically significant than SFM in distinguishing sepsis from systemic inflammatory response syndrome [56]. In general, D-dimer estimates are more readily available in most hospital laboratories and better standardized in view of their usefulness in excluding venous thromboembolic disease [57]. Nonetheless, different commercial assays have different normal ranges, which are not interchangeable. For D-dimer cutoff levels in DIC scoring, we recommend that individual laboratories assign scores for moderate and strong elevation based on 25% and 75% quartiles derived from samples from intensive-care patients [58]. A further consideration is in monitoring D-dimer elevation as accuracy may be an issue, because most assays have been developed for their negative predictive value in excluding venous thromboembolism. Clinicians should also be aware that increasing levels may be from renal and/or liver impairment affecting clearance [59].
Go to :
OTHER MEASUREMENTS
Besides the above-described tests, several other markers have been proposed as useful in diagnosing DIC. Most of these molecular markers, e.g., thrombin-antithrombin (TAT) complexes and prothrombin fragment1+2, are available only in specialized laboratories and even then are not routinely performed and standardized to this setting.
1. Protein C and antithrombin (AT)
Significant reductions in these endogenous anticoagulants correlate with DIC severity and adverse outcomes [606162]. However, their sensitivity for early DIC is unclear as DIC is usually obvious from the global coagulation tests when levels are below 50% of normal [61]. Indeed, worsening trends in PT, platelets, and D-dimer provide sufficient robustness without needing PC or AT levels [39]. Large clinical studies have also shown that it is not PC/AT but PT and D-dimer changes that significantly relate to mortality when analyzed as continuous variables [41].
2. Thromboelastography
With automation of theomboelastography (TEG) to provide point-of-care assessment of coagulation, there is a potential for real-time assessment of a more comprehensive picture of the hemostatic condition [63]. In DIC, Sivula et al [64] identified hypocoagulation in patients with overt DIC, while those without DIC showed hypercoagulation. However, both hyper- and hypocoagulability TEG changes have been linked with adverse clinical events in DIC-relevant diseases [65], and a systematic review of TEG in sepsis confirmed this variability [66]. Often, the values fall within normal reference ranges, and the overall superiority of these tests over conventional tests in DIC has not been proven. Only in cardiac surgery is the use of TEG sufficiently cost and clinically effective. As such, we do not routinely use TEG in DIC and await results from large prospective trials.
3. Potential assays
With the increasing relevance of DAMPs in the cross-talk among coagulation, inflammation, and innate immunity, levels of circulating histones and histone-DNA complexes as well as double-stranded cell-free DNA could have translational relevance in patients with DIC. In 199 patients with suspected DIC, histone–DNA complexes and cell-free DNA levels correlated with the severity of DIC scores [23]. Levels were also significantly higher in nonsurvivors as compared to survivors. As to circulating histones, their direct infusion into mice at high doses induces features of DIC [24]. However, no clinical studies have yet shown their value as diagnostic or prognostic markers in human DIC. Future work is therefore warranted.
Go to :
CONCLUSION
While coagulation activation is its hallmark, our perspective of DIC is that it is indicative of when the host response changes from being protective to becoming maladaptive with potentially lethal consequences. This is because the presence of DIC increases the chances of mortality beyond those of the primary disease. As such, its early recognition with disease-specific targeting provides the best chance of a successful outcome. With its dynamic progression and varying manifestations (Fig. 2), DIC is not a condition whereby the specialist in coagulation can dea with the patient in isolation or remotely from the laboratory. We are a part of a multispecialty clinical team that provides critical care support in decision-making around bleeding and thrombotic risks and benefits from interventions.
Go to :
 XML Download
XML Download