

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Nosocomial transmission of influenza is a recognized problem for vulnerable patients, including pediatric, immunosuppressed, hematologic-oncologic, and elderly patients [123]. Therefore, rapid and accurate identification of an influenza outbreak is essential for patient care and treatment.
Respiratory viral infections are mostly diagnosed on the basis of culture, rapid antigen test, or molecular diagnostic assays. Although molecular diagnostic assays show superior sensitivity than the conventional assays, these are generally designed to identify only certain target viruses [45]. Recently developed next-generation sequencing (NGS) technology can provide deep sequencing in an untargeted manner. Here we demonstrate that NGS-based unbiased deep sequencing can be a useful tool to accurately identify virus subtypes from the clinical specimens in the event of a nosocomial influenza outbreak.
An outbreak of influenza-like illness was suspected in a pulmonary ward of Seoul National University Hospital, Seoul, Korea on Jan 2014. More than 10 individuals, including the ward nurses and patients were evaluated as Flu A positive on the basis of influenza antigen test using a BD Directigen EZ Flu A+B Test kit (BD Diagnostics, Sparks, MD, USA) on 20-22 Jan, 2014. Subsequently, two molecular studies were simultaneously performed for six patients, whose specimens were available: 1) a conventional study using respiratory virus multiplex PCR and seasonal H1/H3 PCR, and 2) an NGS-based whole genome sequencing. For the respiratory virus multiplex PCR, we used the Seeplex RV 12 ACE Detection kit (Seegene, Seoul, Korea) according to the manufacturer's instructions. The seasonal H1/H3 PCR was carried out by using Seeplex FluA ACE Subtyping kit (Seegene). This study was approved by the Institutional Review Board of Seoul National University Hospital.
For whole viral genome sequencing, total RNA was extracted by using QIAamp Viral RNA Mini kit (Qiagen, Hilden, Germany). Double-stranded cDNA was prepared from 5 µg of total RNA by the random priming method using the SuperScript Choice System for cDNA synthesis (Invitrogen, Carlsbad, CA, USA), and processed with TruSeq Stranded Total RNA Sample Prep kit (Illumina Inc., San Diego, CA, USA) according to the manufacturer's instruction. The resulting library was quantified and assessed for its quality by using Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA), and the average library size was 300 bp. This library was sequenced by using Illumina MiSeq (Illumina Inc.) with reagent kit v2 (Illumina Inc.). Sequencing was performed with paired-end reads of 250 bp in length.
Bioinformatic analyses were carried out after the procedures. First, total reads were mapped to the human hg19 reference genome to filter out reads that originated from the human genome, and unmapped reads were collected for further analysis by using a mapping module in CLC Genomics Workbench software version 6.5.1 (CLC bio, Cambridge, MA, USA). Second, the genome assembly was constructed from these unmapped reads by using a de novo module in the same software, with minimum contiguous length set at 200 bp for assembling consensus sequences and the other parameters set at default. Third, the assembled contigs (≥500 bp) were compared against the nucleotide database of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov) by using the BLAST algorithm (http://blast.ncbi.nlm.nih.gov/). The highest scoring BLAST match was filtered according to a minimum 90% identity and 90% query coverage. Then, common BLAST matches were selected for phylogenetic analysis. Finally, assembled contigs and candidate viral reference genome sequences were aligned in ClustalW (http://www.clustal.org), and phylogenetic analysis was performed by using the maximum likelihood method.
The Seeplex RV 12 PCR confirmed influenza A in three of these patients (P1, P4, and P6), but failed to demonstrate influenza A in the remaining three patients (P2, P3, and P5). Seasonal H1 and H3 were excluded for the influenza A positive patients. The molecular study results are summarized in Table 1.
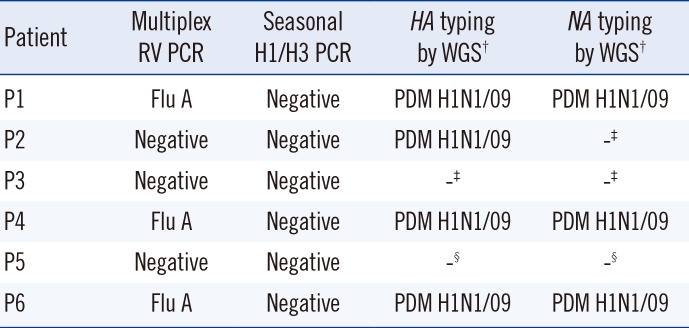
Table 1
Molecular study results for six patients*
![]()
Total of 7 to 12 million reads were obtained from five patients. The sample from patient P5 was excluded from whole-genome sequencing because the extracted RNA quality was poor (Table 2). After mapping to the human reference genome, we further analyzed the 3-4% of total reads that were not identified as originating from a human source.
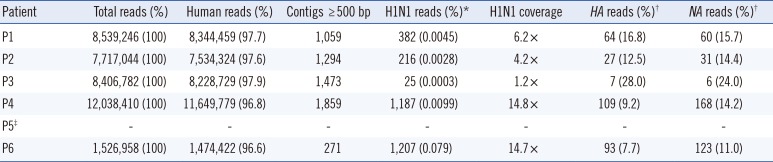
Table 2
Sequencing statistics of the study
*The pandemic H1N1 reference genome used here is the A/Korea/01/2009(H1N1) strain. GenBank accessions are GQ160811-3, GQ131023-6, and GQ132185; †These percentages are for reads only mapped to the pandemic H1N1 reference genome; ‡P5 was excluded from whole-genome sequencing because the quality of extracted RNA was poor.
![]()
With de novo assembly of nonhuman reads, an average of 14,999 contigs per patient was constructed. We selected 5,956 total contigs with a minimum length of 500 bp and performed a BLAST search to identify similar viral genomes (Table 2).
Our BLAST search indicated that most contigs were originated from human and bacterial (e.g., Staphylococcus epidermidis and Pseudomonas spp.) sources. The remaining contigs showed high similarity to pandemic H1N1 virus sequences. The size of these contigs ranged from 524 bp to 2,299 bp (average 844 bp).
Phylogenetic analysis of the HA and NA genes included various influenza A strains like A/Korea/01/2009(H1N1), A/Korea/426/68(H2N2), A/New York/392/2004(H3N2). The analysis showed that sequences from both the HA and NA genes of our samples clustered closely with sequences from the virus strain A/Korea/01/2009(H1N1) (Fig. 1): percent identity between our samples and A/Korea/01/2009(H1N1) was 98.6-99.6% for HA and 99.2-99.5% for NA.
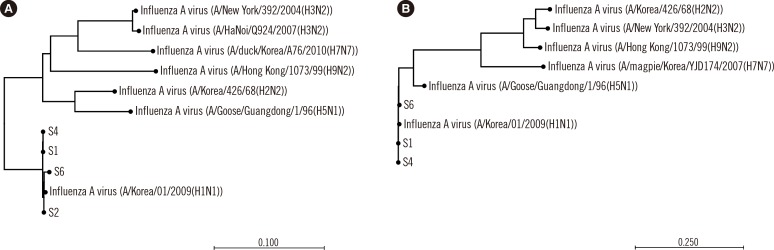
We analyzed the genome coverage of the non-human reads for the influenza A(H1N1)pdm09 virus using A/Korea/01/2009 (H1N1) (GenBank accessions: GQ160811-3, GQ131023-6, GQ 132185), one closely related pandemic H1N1 strain for which the whole genome sequence is available. An average of 603 reads (25-1,207) mapped to this pandemic H1N1 virus and the overall read coverage was 8.2× (1.2×-14.8×) (Table 2). Among the reads mapped to this pandemic virus, 7.7-28.0% reads were mapped to the HA gene, and 11.0-24.0% were mapped to the NA gene.
Whole-genome sequencing of influenza A virus has been used to determine the genetic basis of pathogenicity and antiviral resistance and to identify mixed infections or quasispecies [67]. Whole-genome sequencing has also been applied for various epidemiological investigations, such as outbreaks of neonatal methicillin-resistant Staphylococcus aureus, Mycobacterium tuberculosis, and multi-drug resistant Escherichia coli [8910]. To our knowledge, however, this study is the first to investigate an influenza outbreak by whole-genome sequencing in clinical specimens. Unlike other molecular methods that can detect only a limited number of virus targets, whole-genome sequencing can provide information unbiased by prior knowledge of the viral etiology of an outbreak. In addition, deep sequencing enables us to identify the exact cause of an epidemic event in clinical specimens with viral RNA or DNA in very small quantities, for example, in specimens containing viral targets equivalent to 0.005% of the total sequencing reads as seen in our study.
As well as methodological robustness and cost, the turn-around time (TAT) needs to be considered before whole-genome sequencing is applied to the investigation of nosocomial outbreaks by a diagnostic microbiology laboratory. Sherry et al. [8] reported five days of TAT from a positive culture to the completion of sequencing to investigate putative multidrug-resistant E. coli. In our study, the TAT from library preparation for MiSeq sequencing to completion of sequence analysis was three days: two days for sequencing and one day for sequence analysis. This time is longer than that of other molecular methods, but this time might be improved with the advancement of sequencing technologies. The value of unbiased information at the sequence level, which this approach provides, should also be considered when choosing a method of investigation.
Until now, at least seven phylogenetically distinct viral clades of pandemic H1N1 virus have been identified [11]. The pandemic H1N1 virus identified in our study was most closely related to the A/Wisconsin/11/2013 strain. Among the amino acid changes defining clades 1-7, this virus had unique amino acid substitutions in the NP (V100I), NA (N248D), NS1 (I123V), and HA (S220T) regions. Although there were no substitutions at codon 106 of NA, this virus can be classified as clade 7, the most commonly isolated clade of H1N1 influenza virus in the world [11].
In summary, NGS-based unbiased sequencing can be effectively applied to investigate molecular characteristics of nosocomial influenza outbreak in clinical specimens such as nasopharyngeal swabs.
 XML Download
XML Download