This article has been
cited by other articles in ScienceCentral.
Dear Editor,
High-throughput and cost-effective
BRCA genetic screening is needed for application of pharmacogenetics in personalized HBOC therapy. Banerjee et al. [
1] suggested that poly(ADP-ribose) polymerase (PARP) inhibitors show considerable promise for the treatment of cancers with
BRCA1 or
BRCA2 mutations. Secord et al. [
2] demonstrated that compared to the global use of PARP inhibition, the
BRCA1 and
BRCA2 test for personalized PARP inhibition treatment may represent a de facto cost-reducing strategy.
In this study, results of targeted Next-generation sequencing (NGS) by Ion Torrent Personal Genome Machine (PGM, Life Technologies, Guilford, CT, USA) were compared with those of Sanger sequencing in seven HBOC patients harboring pathogenic variant or rare variant of BRCA1 and BRCA2 of uncertain clinical significance.
All enrolled subjects provided written informed consent for clinical and molecular analyses, and the study protocol was approved by the institutional review board (KC15SISE0263) of The Catholic University, Seoul, Korea. Seven HBOC patients harboring pathogenic or unclassified variants of
BRCA1 and
BRCA2 as confirmed by Sanger sequencing were studied. The Ion AmpliSeq
BRCA1 and
BRCA2 Panel (Life Technologies) consisting of 167 primer pairs in three primer pair pools was used for targeted NGS analysis. Sequencing on the Ion Torrent PGM was performed by using 500 flow runs that generated approximately 200 bp reads. Torrent Variant Caller 3.4 was applied for alignment and variant detection. The variant caller parameter setting was germline PGM high stringency (
Table 1). Sequence data was visually confirmed with the Integrative Genomics Viewer (IGV) and any sequence, alignment, or variant call error artifacts were discarded. Non-synonymous variants called were evaluated by using ClinVar (
http://www.ncbi.nlm.nih.gov/clinvar), the BIC database (
http://research.nhgri.nih.gov/bic/), and the HGMD database (
http://www.hgmd.cf.ac.uk/). Minor allele frequency (MAF) was determined from the 1000 Genomes Project database (
http://www.1000genomes.org/).
Technical performance of the Ion AmpliSeq BRCA1/2 Panel showed 81% of template-positive ion sphere particle sample loading. The total read obtained was 4,541,406, with a mean read length of 137 bp. The mean sequencing depth for each region was ×494 and the average uniformity of coverage was 99%, which is percentage of target bases, covered by at least ×0.2 of the average base read depth. All variants located in all coding exons and in adjacent intronic regions of BRCA1 and BRCA2 identified by Sanger sequencing were detected by targeted NGS analysis. Direct sequencing of entire coding exons and flanking intronic sequences of BRCA1 and BRCA2 was performed on ABI 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems).
Of seven patients, four carried deleterious
BRCA1 or
BRCA2 variants: two frameshift and two nonsense mutations. The p.Asn567Ilefs
*5 and p.Arg1203
* of BRCA1 have previously been reported in Turkish [
3] and American [
4] populations, respectively (
Fig. 1A, B). The p.Ser1248Argfs
*10 and p.Arg2494
* of BRCA2 have been reported in German [
5] and Finnish [
6] populations, respectively (
Fig. 1C, D). Meanwhile two
BRCA1 and one
BRCA2 out of nine non-synonymous variants are rare according to 1,000 Genome Project data (<1% population MAF). The p.Leu52Phe of BRCA1 and p.Val2109Ile of BRCA2 have been reported in the Koreans, and the p.Tyr856His of BRCA1 has been found in the Japanese populations.
Of various NGS platforms, the PGM generates DNA sequencing reads by detecting ions released when deoxynucleotide triphosphates are incorporated into a growing DNA strand on a semiconductor device [
7]. In particular, it has been documented that indel errors occurring in homopolymer DNA regions significantly affect the specificity of indel detection owing to the nature of sequencing chemistry of PGM [
8]. In this study, sequence "AGTG" at position 3972_3975 of
BRCA2 was given in NGS; however, the Human Genome Variation Society notation prescribes that on the forward strand it should be "TGAG" at position 3974_3977 as given in Sanger sequencing.
While the protein-truncating variants (either frameshift, nonsense, or splice) are located generally in the coding exons or the flanking intronic sequences of
BRCA genes, potentially deleterious alterations may also reside in the noncoding intronic sequences. For example, deep intronic mutation causing activation of a cryptic exon in
BRCA2 has been reported in a French family with a history of breast cancer [
9]. Thus, rare variants called with both NGS and Sanger sequencing should be considered for validation study, regardless of their location.
In conclusion, our study is a clear example that the quality of targeted NGS of a disease-specific subset of genes is equal to the quality of Sanger sequencing, and therefore it can be implemented reliably as a stand-alone diagnostic test demonstrated by Sikkema-Raddatz B et al. [
10].
Acknowledgments
We are grateful to The Catholic Genetic Laboratory Center for assisting us in carrying out this study and compiling this report. This research was supported by a grant of the Korea Healthcare technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (HI14C3417).
References
1. Banerjee S, Kaye SB, Ashworth A. Making the best of PARP inhibitors in ovarian cancer. Nat Rev Clin Oncol. 2010; 7:508–519. PMID:
20700108.

2. Secord AA, Barnett JC, Ledermann JA, Peterson BL, Myers ER, Havrilesky LJ. Cost-effectiveness of BRCA1 and BRCA2 mutation testing to target PARP inhibitor use in platinum-sensitive recurrent ovarian cancer. Int J Gynecol Cancer. 2013; 23:846–852. PMID:
23666017.
3. Yazici H, Glendon G, Yazici H, Burnie SJ, Saip P, Buyru F, et al. BRCA1 and BRCA2 mutations in Turkish familial and non-familial ovarian cancer patients: a high incidence of mutations in non-familial cases. Hum Mutat. 2002; 20:28–34. PMID:
12112655.
4. Friedman LS, Ostermeyer EA, Szabo CI, Dowd P, Lynch ED, Rowell SE, et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet. 1994; 8:399–404. PMID:
7894493.
5. Meindl A. Comprehensive analysis of 989 patients with breast or ovarian cancer provides BRCA1 and BRCA2 mutation profiles and frequencies for the German population. Int J Cancer. 2002; 97:472–480. PMID:
11802209.
6. Vehmanen P, Friedman LS, Eerola H, McClure M, Ward B, Sarantaus L, et al. Low proportion of BRCA1 and BRCA2 mutations in Finnish breast cancer families: evidence for additional susceptibility genes. Hum Mol Genet. 1997; 6:2309–2315. PMID:
9361038.
7. Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011; 475:348–352. PMID:
21776081.
8. Bragg LM, Stone G, Butler MK, Hugenholtz P, Tyson GW. Shining a light on dark sequencing: characterising errors in Ion Torrent PGM data. PLoS Comput Biol. 2013; 9:e1003031. PMID:
23592973.
9. Anczuków O, Buisson M, Léoné M, Coutanson C, Lasset C, Calender A, et al. BRCA2 deep intronic mutation causing activation of a cryptic exon: opening toward a new preventive therapeutic strategy. Clin Cancer Res. 2012; 18:4903–4909. PMID:
22753590.
10. Sikkema-Raddatz B, Johansson LF, de Boer EN, Almomani R, Boven LG, van den Berg MP, et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat. 2013; 34:1035–1042. PMID:
23568810.
Fig. 1
Identification of deleterious mutations in BRCA1 or BRCA2 by Sanger sequencing and next-generation sequencing (NGS), as visualized by Sequencher Software (top) and Integrative Genomics Viewer (IGV, bottom), respectively. The deletion(s) or base change is indicated by a red arrow in Sanger sequencing and is represented by a black dashes in IGV. (A) A deletion A at position 1700 of cDNA (c.1700delA; p.Asn567Ilefs*5) of BRCA1 in patient hereditary breast and/or ovarian cancer (HBOC)1; (B) A substitution C to T at position 3607 of cDNA (c.3607C>T; p.Arg1203*) of BRCA1 in patients HBOC3; (C) A deletion TGAG at position 3744-3747 of cDNA (c.3744_3747delTGAG; p.Ser1248Argfs*10) of BRCA2 in patient HBOC5; (D) A substitution C to T at position 7480 of cDNA (c.7480C>T; p.Arg2494*) of BRCA2 in patients HBOC6.
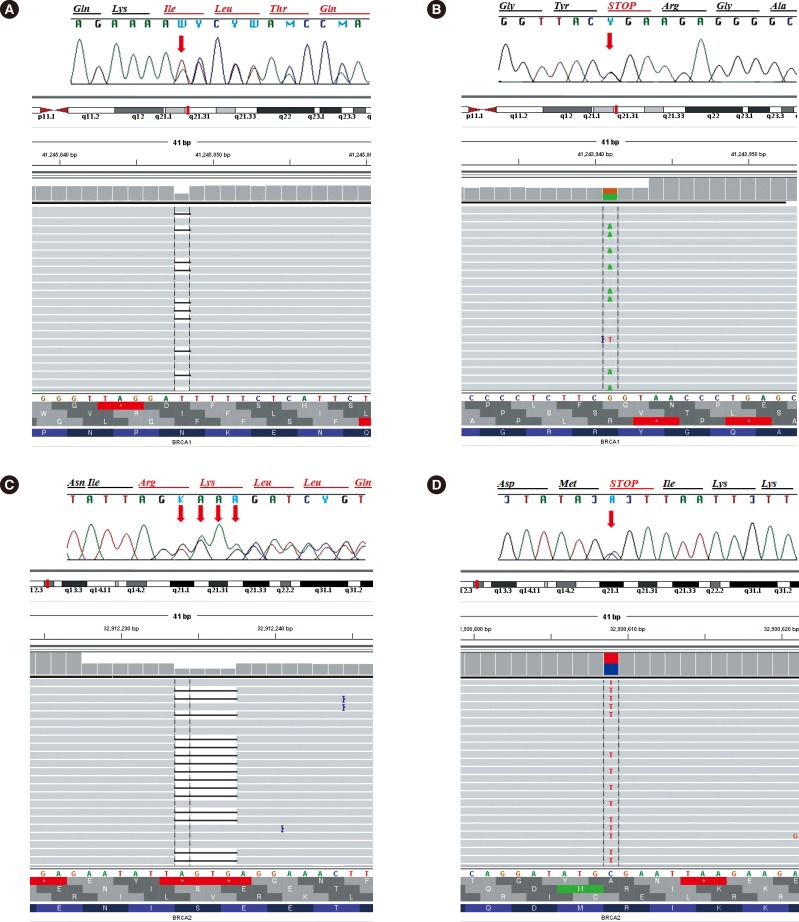
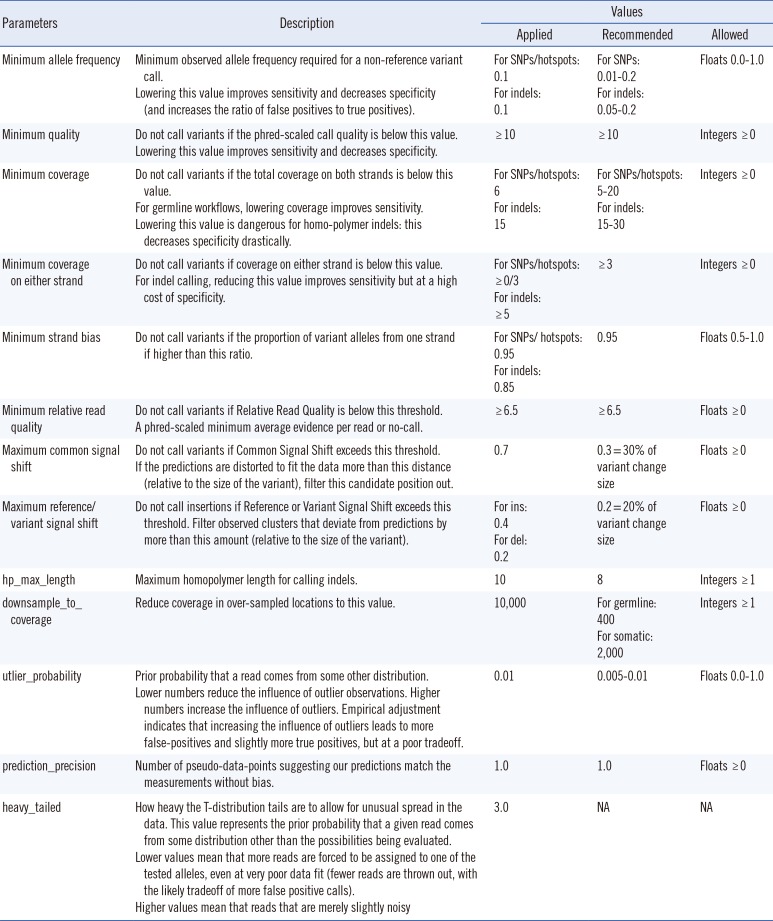
Table 1
Override parameters selected for customizing hotspot calling of BRCA1 and BRCA2 variants in this study
|
Parameters |
Description |
Values |
|
Applied |
Recommended |
Allowed |
|
Minimum allele frequency |
Minimum observed allele frequency required for a non-reference variant call. |
For SNPs/hotspots: 0.1 |
For SNPs: 0.01-0.2 |
Floats 0.0-1.0 |
|
Lowering this value improves sensitivity and decreases specificity (and increases the ratio of false positives to true positives). |
For indels: 0.1 |
For indels: 0.05-0.2 |
|
Minimum quality |
Do not call variants if the phred-scaled call quality is below this value. |
≥ 10 |
≥ 10 |
Integers ≥ 0 |
|
Lowering this value improves sensitivity and decreases specificity. |
|
Minimum coverage |
Do not call variants if the total coverage on both strands is below this value. |
For SNPs/hotspots: 6 |
For SNPs/hotspots: 5-20 |
Integers ≥ 0 |
|
For germline workflows, lowering coverage improves sensitivity. |
For indels: 15 |
For indels: 15-30 |
|
Lowering this value is dangerous for homo-polymer indels: this decreases specificity drastically. |
|
Minimum coverage on either strand |
Do not call variants if coverage on either strand is below this value. |
For SNPs/hotspots: ≥ 0/3 |
≥3 |
Integers ≥ 0 |
|
For indel calling, reducing this value improves sensitivity but at a high cost of specificity. |
For indels: ≥5 |
|
Minimum strand bias |
Do not call variants if the proportion of variant alleles from one strand if higher than this ratio. |
For SNPs/hotspots: 0.95 |
0.95 |
Floats 0.5-1.0 |
|
For indels: 0.85 |
|
Minimum relative read quality |
Do not call variants if Relative Read Quality is below this threshold. |
≥ 6.5 |
≥ 6.5 |
Floats ≥ 0 |
|
A phred-scaled minimum average evidence per read or no-call. |
|
Maximum common signal shift |
Do not call variants if Common Signal Shift exceeds this threshold. |
0.7 |
0.3 = 30% of variant change size |
Floats ≥ 0 |
|
If the predictions are distorted to fit the data more than this distance (relative to the size of the variant), filter this candidate position out. |
|
Maximum reference/variant signal shift |
Do not call insertions if Reference or Variant Signal Shift exceeds this threshold. Filter observed clusters that deviate from predictions by more than this amount (relative to the size of the variant). |
For ins: 0.4 |
0.2 = 20% of variant change size |
Floats ≥ 0 |
|
For del: 0.2 |
|
hp_max_length |
Maximum homopolymer length for calling indels. |
10 |
8 |
Integers ≥ 1 |
|
downsample_to_coverage |
Reduce coverage in over-sampled locations to this value. |
10,000 |
For germline: 400 |
Integers ≥ 1 |
|
For somatic: 2,000 |
|
utlier_probability |
Prior probability that a read comes from some other distribution. |
0.01 |
0.005-0.01 |
Floats 0.0-1.0 |
|
Lower numbers reduce the influence of outlier observations. Higher numbers increase the influence of outliers. Empirical adjustment indicates that increasing the influence of outliers leads to more false-positives and slightly more true positives, but at a poor tradeoff. |
|
prediction_precision |
Number of pseudo-data-points suggesting our predictions match the measurements without bias. |
1.0 |
1.0 |
Floats ≥ 0 |
|
heavy_tailed |
How heavy the T-distribution tails are to allow for unusual spread in the data. This value represents the prior probability that a given read comes from some distribution other than the possibilities being evaluated. |
3.0 |
NA |
NA |
|
Lower values mean that more reads are forced to be assigned to one of the tested alleles, even at very poor data fit (fewer reads are thrown out, with the likely tradeoff of more false positive calls). |
|
Higher values mean that reads that are merely slightly noisy |


 PDF
PDF ePub
ePub Citation
Citation Print
Print


 XML Download
XML Download