

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Deficiencies of coagulation factors such as fibrinogen and factors II, V, V + VIII, VII, X, XI, and XIII are called rare bleeding disorders (RBDs). Congenital factor V (FV) deficiency is caused by mutations in F5, which is located on chromosome 1q24, and accounts for about 8% of all RBDs. FV is important for modulating thrombin production. Organization of FV is highly homologous to that of factor VIII. Clinical manifestations of FV deficiency (FVD) are diverse, ranging from life threatening to asymptomatic, even among patients with severe deficiency. FVD is suspected by prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT) that are corrected on mixing tests and is diagnosed by a significantly decreased FV activity. We described genetically confirmed FVD cases in two unrelated Korean families and reviewed previous cases of FVD in Korea.
Proband 1 was a 14-yr-old girl with a history of hematuria and menorrhagia. She had prolonged PT/aPTT and decreased FV activity of 41% (Table 1). Her asymptomatic mother had prolonged PT/aPTT and FV activity of 44%. Her father yielded normal results for coagulation tests. Proband 2 was a 51-yr-old woman with easy bruisability. She had prolonged PT/aPTT and FV activity of 3%. Her daughter had normal PT/aPTT and FV activity of 39%. The daughter reported pain in the left hip, which was suspected to be caused by a hematoma according to ultrasound examination. Both probands yielded normal results for other coagulation factors. Genomic DNA was extracted from the peripheral blood of probands and family members after obtaining their written informed consent. Direct sequencing of F5 identified causative mutations in both probands. Proband 1 was heterozygous for missense mutation c.286G>C (p.Asp96His; Fig. 1). Proband 2 was compound heterozygous for in-frame deletion c.6027_6032delGAACAG (p.Asn2010_Ser2011del) and missense mutation c.6604C>T (p.Arg2202Cys; Fig. 1). A review of the literature and databases showed that the above three F5 mutations had been reported previously [1234]. The mother of proband 1 was heterozygous for Asp96His, and the daughter of proband 2 was heterozygous for Asn2010_Ser2011del (Table 1).
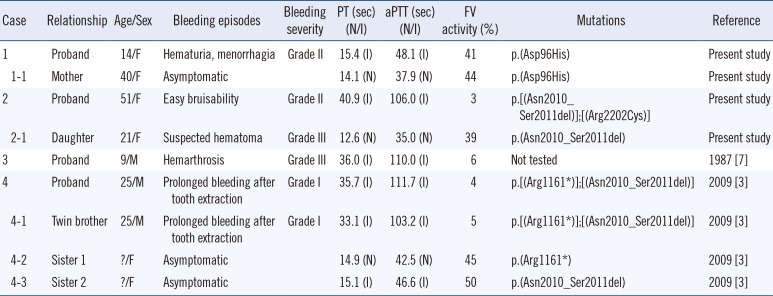
 | Fig. 1(A) Molecular analyses of F5 in two unrelated Koreans patients with congenital FV deficiency. Proband 1 and her mother were heterozygous for c.286G>C (p.Asp96His) (upper panel). Proband 2 was compound heterozygous for c.6027_6032del GAACAG (p.Asn2010_Ser2011del) and c.6604C>T (p.Arg2202Cys), and her daughter was heterozygous for c.6027_6032 delGAACAG (p.Asn2010_Ser2011del) (lower panel). The arrows indicate the mutations.(B) Comparative genomic analyses by using the CLUSTAL W algorithm showed that the Arg2202 residue, which corresponds to the Arg2326 residue of factor VIII, is highly conserved across mammalian species (indicated by an arrow).
|
Table 1
Clinical features and laboratory findings of patients with congenital factor V deficiency in Korea
| Case | Relationship | Age/Sex | Bleeding episodes | Bleeding severity | PT (sec) (N/I) | aPTT (sec) (N/I) | FV activity (%) | Mutations | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Proband | 14/F | Hematuria, menorrhagia | Grade II | 15.4 (I) | 48.1 (I) | 41 | p.(Asp96His) | Present study |
| 1-1 | Mother | 40/F | Asymptomatic | 14.1 (N) | 37.9 (N) | 44 | p.(Asp96His) | Present study | |
| 2 | Proband | 51/F | Easy bruisability | Grade II | 40.9 (I) | 106.0 (I) | 3 | p.[(Asn2010_Ser2011del)];[(Arg2202Cys)] | Present study |
| 2-1 | Daughter | 21/F | Suspected hematoma | Grade III | 12.6 (N) | 35.0 (N) | 39 | p.(Asn2010_Ser2011del) | Present study |
| 3 | Proband | 9/M | Hemarthrosis | Grade III | 36.0 (I) | 110.0 (I) | 6 | Not tested | 1987 [7] |
| 4 | Proband | 25/M | Prolonged bleeding after tooth extraction | Grade I | 35.7 (I) | 111.7 (I) | 4 | p.[(Arg1161*)];[(Asn2010_Ser2011del)] | 2009 [3] |
| 4-1 | Twin brother | 25/M | Prolonged bleeding after tooth extraction | Grade I | 33.1 (I) | 103.2 (I) | 5 | p.[(Arg1161*)];[(Asn2010_Ser2011del)] | 2009 [3] |
| 4-2 | Sister 1 | ?/F | Asymptomatic | 14.9 (N) | 42.5 (N) | 45 | p.(Arg1161*) | 2009 [3] | |
| 4-3 | Sister 2 | ?/F | Asymptomatic | 15.1 (I) | 46.6 (I) | 50 | p.(Asn2010_Ser2011del) | 2009 [3] |
![]()
At present, more than 100 mutations have been identified in F5 (Human Gene Mutation Database professional version, release 2015.1; last access, April 13, 2015), of which 50% are missense mutations [5]. Most missense mutations in F5 are clustered in A and C2 domains. Asp96His and Arg2202Cys in the present study are located in A1 and C2 domains, respectively. Asp96His (Asp68His in the mature protein) was previously described in four unrelated Chinese patients with FVD and was caused by the disruption of salt bridge and subsequent interference in protein expression [12]. Arg2202Cys was reported in a Chinese patient [2]. Arg2202 residue of FV was highly conserved across mammalian species (Fig. 1B). Although Arg2202Cys was predicted to be mutational, it was not detected in 100 control chromosomes of Korean descent. Asn2010_Ser2011 are located in a loop on the surface of C1 domain and are in close contact with another loop of A3 domain, and Asn2010_Ser2011del was reported in a Korean patient [3]. Asn2010_Ser2011del destabilizes C1-A3 interaction, thus affecting the folding, stability, and secretion of FV [3]. Thus, detection of these F5 mutations in Asian patients suggests shared genetic backgrounds among Asian ethnicities [1234].
The 2013 Annual Report of Korea Hemophilia Foundation indicated that RBDs, including FVD (0.2%), represented about 4% of all bleeding disorders in Korea [6]. Except the previous cases reported by Song et al., [3] a literature review revealed that previous Korean case of FVD lacked genetic information (Table 1) [7]. The European Network of Rare Bleeding Disorders recently established categories of bleeding severity, ranging from asymptomatic to grades I, II, and III [8]. The association between clinical severity and coagulation factor activities has been variably reported in RBDs, with a poor association for FVD [9]. A recent study explained this poor association by highlighting the important role of FV in platelet compartments [10]. In this study, proband 1 showed grade II, while her mother was asymptomatic, despite showing similar FV activity from the same mutation (intrafamilial variability). Proband 2 showed grade II, with 3% FV activity from biallelic mutations, while her daughter was suspected of having grade III, with 39% FV activity from a monoallelic mutation. In contrast, a family member of a previously reported patient who had the same genotype as the daughter of the proband 2 was asymptomatic. This underscores the importance of careful analysis of genotype-phenotype correlations in patients with FVD.
 XML Download
XML Download