INTRODUCTION
Congenital hypothyroidism (CH) is one of the most common causes of preventable mental retardation, and it is routinely screened worldwide during the newborn period. Before the introduction of newborn screening (NBS), the incidence of CH was 1 in 6,500 live births, whereas the incidence was initially reported to be approximately 1 in 3,000 newborns with NBS [
1]. With more experience from the NBS program, the incidence of CH has been reported to vary considerably among ethnic groups [
2345]. A recent study showed that the overall incidence of CH was 1 in 1,706 in a pan-ethnic group [
2].
Compared with the incidence estimated by NBS tests, there are scarce data regarding the incidence of CH associated with mutations in the population. Only a few studies regarding the genetic incidence of CH have been published [
678910]. For instance, the incidence of
TPO mutations has been reported to be 1 in 66,000 in the Dutch population, 1 in 177,000 in the Japanese population, and 1 in 20,000 in the Slovakian population [
678]. Further studies in the Japanese populations revealed that the incidence of
TSHR mutations,
DUOX2 mutations, and
PAX8 mutations was estimated to be 1 in 118,000, 1 in 44,000, and 1 in 176,000, respectively [
910]. These studies suggested that there are ethnic differences in the incidence of CH owing to mutations and that there is genetic heterogeneity underlying CH. Little is known about the genetic incidence of CH in the Korean population. There were only two previous studies regarding genetics of CH in Korean patients with CH [
1112]. One study in the Korean patients with thyroid dyshormonogenesis (DH) showed a considerably lower rate of mutation positivity in
TPO mutations [
11]. Another study in the Korean patients with CH found that
DUOX2 mutations are a common cause of CH with eutopic thyroid glands [
12]. The two previous studies were based on a relatively small series of Korean patients from hospital setting. Until now, little is known on the mutations underlying CH in the Korean population.
Another important consideration is the classification criteria to estimate the incidence of CH. On the basis of the current classification, CH encompasses very distinct morphologic phenotypes, including thyroid dysgenesis (TD) and DH. TD is present in 80-85% of patients, whereas DH accounts for the remaining 15-20% of patients. However, this classification system also includes patients belonging to an intermediate category-those with normal results on thyroid scintigraphy (neither TD nor DH). Exclusion of this intermediate group might influence the variable incidence rate of CH in different populations. The previous genetic epidemiology studies were performed according to a phenotype-first approach (TD or DH) by using this classification [
678910]. Thus, the true genetic incidence of CH remains elusive. To estimate the real incidence of CH associated with mutations, it is necessary to include this intermediate group.
Although TD is usually a sporadic condition, several genes, including
TSHR and
PAX8, have been implicated in TD. In contrast, other genes, such as
PDS,
TPO,
TG,
DUOX2,
DUOXA2, and
SCL5A5 are involved in thyroid hormone synthesis and secretion (DH). In general, it is expected that DH is inherited in an autosomal recessive manner. However, recent studies have reported notable exceptions and complex genotype-phenotype correlations [
1213141516171819]. For instance, biallelic and triallelic mutations in
DUOX2 cause permanent CH, whereas a monoallelic mutation also suffices to cause transient CH [
1420]. In addition, environmental factors such as iodine excess could delay the onset of CH with biallelic mutations in
DUOX2 [
19]. Occasionally, monoallelic mutations of
TPO,
PAX8, or
TSHR might result in transient CH [
71718]. Interestingly, another study reported that the coexistence of a monoallelic
TPO mutation and heterozygous mutations in
TSHR in the same individual does not influence the severity of the phenotype [
15].
We conducted a population-based study regarding the genetic screening of CH directly detected on NBS. This study aimed to determine the incidence of CH associated with mutations in the Korean newborn population. In addition, we evaluated the mutation frequency and spectrum underlying CH and investigated whether there was a genotype-phenotype correlation using the data from outpatients.
Go to :

METHODS
1. Subjects
We selected subjects from two different situations to evaluate the genetic aspects of CH. In the first situation, genetic screening was conducted in all the newborns with thyroid dysfunction detected on current NBS. We chose this approach in an attempt to include the intermediate group of CH (neither TD nor DH). A total of 112 newborns with thyroid dysfunction were enrolled from 256,624 consecutive NBS at the Seoul Medical Science Institute in Korea from January 2011 to May 2014. The newborn population in this study represented approximately 18% of births (256,624/1,428,400) in Korea during the designated time.
The level of thyroid-stimulating hormone (TSH) was measured in dried blood spots (DBS) by using an AutoDELFIA Neonatal hTSH kit (PerkinElmer, Waltham, MA, USA) on postnatal days 3-5. A total of 1,518 newborns with TSH levels higher than 10 mU/L on the primary screening tests were recalled, and 1,190 of those newborns were retested by using second samples. Thyroid dysfunction was defined as a positive NBS with abnormal TSH levels in the two separate samples. A total of 200 newborns were found to have thyroid dysfunction under these criteria, and ultimately, 112 newborns had a DBS with an adequate amount of DNA to extract for our study. The researchers were blinded to all information regarding the identification of the newborns.
In the second situation, genetic screening was performed in 58 outpatients with confirmed primary CH. They were followed up owing to their primary CH from 2009 to 2014 in an endocrine clinic in the Samsung Medical Center in Korea. For the outpatients, we collected the following information: demographic data, prematurity history, associated anomalies, and the results of the Tc-99m thyroid scan and thyroid ultrasound. This study was approved by the Institutional Review Boards at the Seoul Medical Science Institute and Samsung Medical Center; informed consent from enrolled patients was exempt for this retrospective study that use stored biospecimens.
2. Genetic screening
All DBS were stored at -20℃ until the extraction of genomic DNA. Several 3-mm discs from residual DBS were collected by using a Delfia DBS puncher (PerkinElmer). The discs were incubated overnight at 37℃ in nuclei lysis buffer (500 µL) and proteinase K (15 µL). After protein precipitation, the tube was centrifuged at 11,400 g for 10 min. The supernatant was transferred to a new tube, and isopropanol (500 µL) was added. After centrifugation at 11,400 g for 2 min, the supernatant was discarded, and 70% ethanol (500 µL) was added. After an additional centrifugation at 11,400 g for 2 min, the pellet was air-dried for 15 min. The concentration of extracted DNA was measured by using the Pico-Green dsDNA assay (Quanti-iTTMPicoGreen dsDNA kit; Invitrogen, Carlsbad, CA, USA).
A customized multiplex PCR was applied for 97 genes related to inherited metabolic diseases using the Ion AmpliSeq designer software (Life Technologies, Carlsbad, CA, USA) (manuscript in preparation). Among them, six genes, including genes associated with TD (TSHR and PAX8) and DH (TPO, DUOX2, DUOXA2, and SLC5A5), were selected for genetic screening of CH. All coding exons, intron sequences, and untranslated regions (UTR) of 20-bp flanking each exon were targeted and designed. Regions not covered by the primers among the six genes relevant to CH were 400 bp long, and there were no known mutations found in the Human Genome Mutation Database (HGMD, professional version 2014.01). Targeted sequencing was performed on an Ion PGM sequencer or Ion Proton sequencer (Life Technologies), following the manufacturer's instructions. Sixteen or 20 barcoded samples were sequenced on an Ion 318 chip, and 32 barcoded samples were sequenced on a Proton 1 chip. A total of 170 samples from 112 newborns and 58 outpatients were sequenced.
3. Bioinformatics analysis and mutation prioritization
Data from targeted sequencing were analyzed by using Torrent Suite software (version 4.0.3; Life Technologies). The read was aligned to hg19, and variant calling was performed by using the "Germ Line-PGM-High Stringency" setting. Called variants were functionally annotated by using the ANNOVAR tool [
2122].
To select the mutations, we applied the following criteria: variants with minor allele frequency (MAF) <0.01 in the 1000 Genome Project (
http://browser.1000genomes.org/index.html), the Exome Sequencing Project (
http://evs.gs.washington.edu/EVS/), the Exome Aggregation Consortium (
http://exac.broadinstitute.org/), and the Korean Reference Genome Database (
http://152.99.75.168/KRGDB/); variants predicted to be "deleterious" or "damaging" from Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping v2 (Polyphen-2), likelihood ratio test (LRT), MutationTaster, MutationAssesor, or FATHMM and with a Genomic Evolutionary Rate Profiling (GERP) score higher than 2; removal of variants located in 3'-UTR, 5'-UTR, downstream, upstream, and noncoding RNAs; and the removal of synonymous variants and selection of missense and nonsense mutations in coding sequences, and splicing mutations. Lastly, the HGMD (professional version 2014.01) was searched to evaluate whether the mutations were novel or known.
4. Sanger sequencing
All prioritized mutations were amplified by PCR using primer pairs designed with Primer3 software. Relevant exons and their flanking intron regions were sequenced by using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA) and an ABI Prism 3100 Genetic Analyzer (Applied Biosystems). In case of novel mutations, targeted Sanger sequencing was performed by using 60 healthy individuals as controls.
5. Statistical analyses
The Mann-Whitney U test and Kruskal-Wallis test were used to evaluate the association between TSH level and mutations. The allelic status of the mutations between the newborns and patients was compared with Fisher's exact test. The statistical significance was analyzed with MedCalc version 11.5.1.0 (Mariakerke, Belgium). P values less than 0.05 were considered statistically significant.
Go to :
RESULTS
1. Statistical data of targeted sequencing
The numbers of mapped reads for Ion PGM and Ion Proton were 212,552 and 1,229,961, respectively. The percentage of on-target sequences in each sample showed a median of 90-95%. The median depth of coverage was 92-fold for Ion PGM and 383-fold for Ion Proton, and 99% and 93% of bases showed at least 1-fold and 20-fold coverage, respectively. A total of 1,204 variants were called in 112 newborns. In TSHR, PAX8, TPO, DUOX2, DUOXA2, and SCL5A5, 138 variants were selected. After functional filtering, 47 mutations were prioritized. In the 58 outpatient cases, 28 mutations were prioritized among 1,272 called variants.
2. Incidence
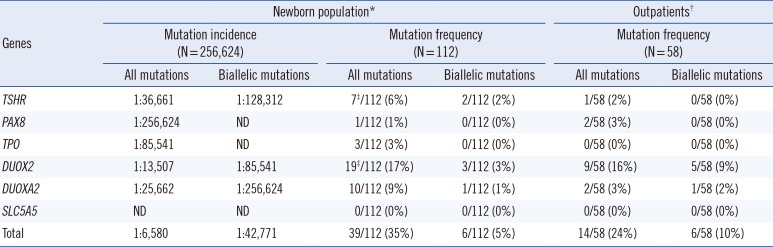
We estimated the incidence of CH based on the current NBS tests to be 1 in 1,283 in the Korean population. The mutation incidence of CH was estimated to be 1 in 6,580 (39/256,624) in the Korean newborn population, according to the mutation status of the six selected genes (
Table 1). When limited to biallelic mutations, the mutation incidence of CH was estimated to be 1 in 42,771 (
Table 1).
Table 1
Mutation incidence and frequency
|
Genes |
Newborn population*
|
Outpatients†
|
|
Mutation incidence (N=256,624) |
Mutation frequency (N=112) |
Mutation frequency (N=58) |
|
All mutations |
Biallelic mutations |
All mutations |
Biallelic mutations |
All mutations |
Biallelic mutations |
|
TSHR
|
1:36,661 |
1:128,312 |
7‡/112 (6%) |
2/112 (2%) |
1/58 (2%) |
0/58 (0%) |
|
PAX8
|
1:256,624 |
ND |
1/112 (1%) |
0/112 (0%) |
2/58 (3%) |
0/58 (0%) |
|
TPO
|
1:85,541 |
ND |
3/112 (3%) |
0/112 (0%) |
0/58 (0%) |
0/58 (0%) |
|
DUOX2
|
1:13,507 |
1:85,541 |
19‡/112 (17%) |
3/112 (3%) |
9/58 (16%) |
5/58 (9%) |
|
DUOXA2
|
1:25,662 |
1:256,624 |
10/112 (9%) |
1/112 (1%) |
2/58 (3%) |
1/58 (2%) |
|
SLC5A5
|
ND |
ND |
0/112 (0%) |
0/112 (0%) |
0/58 (0%) |
0/58 (0%) |
|
Total |
1:6,580 |
1:42,771 |
39/112 (35%) |
6/112 (5%) |
14/58 (24%) |
6/58 (10%) |

3. Frequency and spectrum of pathogenic variants
Thirty-six different mutations were validated in 53 cases (31%, 53/170), which included 35% of the newborn group (39/112) and 24% of the outpatient group (14/58) (
Table 1). Biallelic mutations and monoallelic mutations were identified in 7% (12/170) and 24% (41/170) of the cases, respectively (
Table 1). There was a trend that biallelic inactivation was more frequently observed in outpatients than in newborns (10% vs. 5%), although the association was not significant (
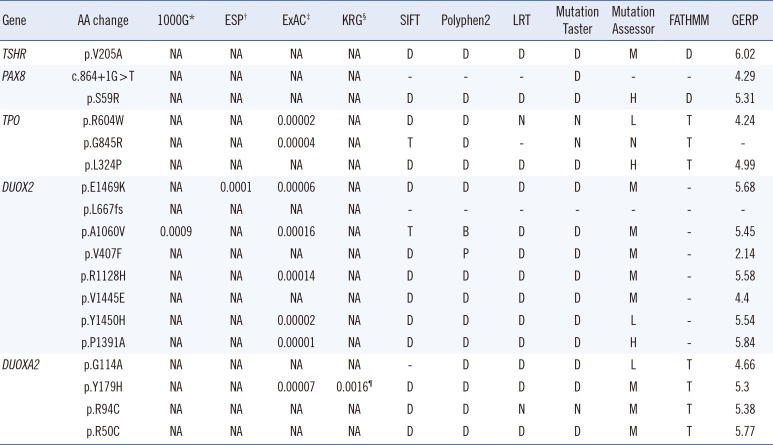
P=0.059). The mutations consisted of 18 novel mutations and 18 known mutations (
Table 2). All of the novel mutations were predicted to have highly deleterious or damaging effects based on the results of several
in silico analyses (
Table 3). On the basis of control databases, such as the 1000 Genome Project, the Exome Sequencing Project, the Exome Aggregation Consortium, and the Korean Reference Genome Database, all of the novel mutant alleles were found to be very rare (
Table 3). The relative proportions of mutated genes between the newborns and outpatients were considerably similar; inactivation mutations in
DUOX2 were the most common in both groups (19/112 in newborns and 9/58 in outpatients,
Table 2 and
Table 3).
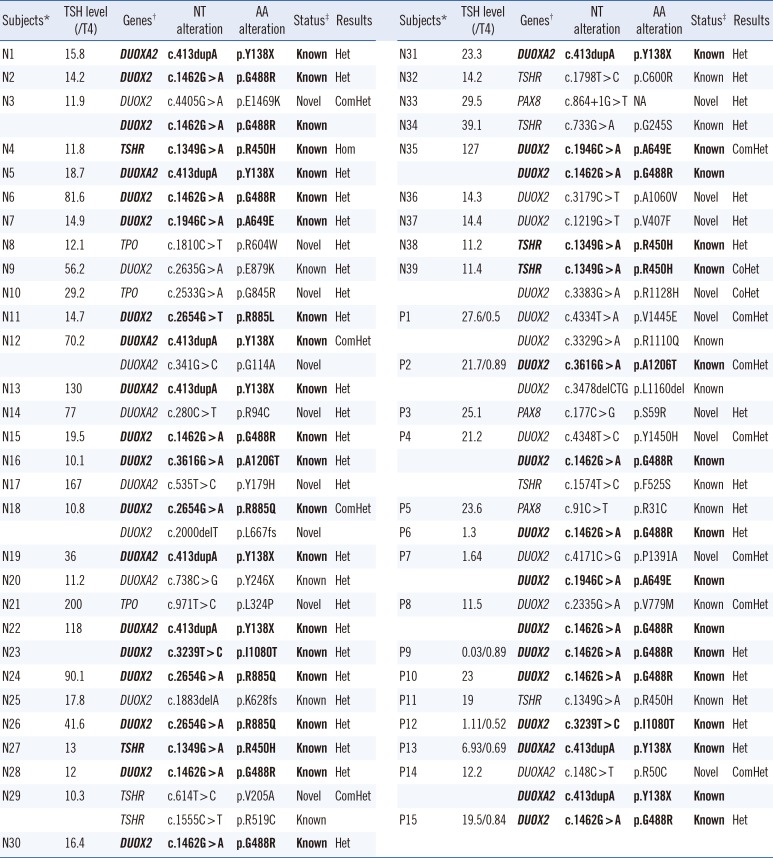
Table 2
Spectrum of mutations identified in this study
|
Subjects*
|
TSH level (/T4) |
Genes†
|
NT alteration |
AA alteration |
Status‡
|
Results |
|
Subjects*
|
TSH level (/T4) |
Genes†
|
NT alteration |
AA alteration |
Status‡
|
Results |
|
N1 |
15.8 |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
Het |
|
N31 |
23.3 |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
Het |
|
N2 |
14.2 |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Het |
|
N32 |
14.2 |
TSHR
|
c.1798T > C |
p.C600R |
Known |
Het |
|
N3 |
11.9 |
DUOX2
|
c.4405G > A |
p.E1469K |
Novel |
ComHet |
|
N33 |
29.5 |
PAX8
|
c.864+1G > T |
NA |
Novel |
Het |
|
|
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
|
|
N34 |
39.1 |
TSHR
|
c.733G > A |
p.G245S |
Known |
Het |
|
N4 |
11.8 |
TSHR
|
c.1349G > A
|
p.R450H
|
Known
|
Hom |
|
N35 |
127 |
DUOX2
|
c.1946C > A
|
p.A649E
|
Known
|
ComHet |
|
N5 |
18.7 |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
Het |
|
|
|
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
|
|
N6 |
81.6 |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Het |
|
N36 |
14.3 |
DUOX2
|
c.3179C > T |
p.A1060V |
Novel |
Het |
|
N7 |
14.9 |
DUOX2
|
c.1946C > A
|
p.A649E
|
Known
|
Het |
|
N37 |
14.4 |
DUOX2
|
c.1219G > T |
p.V407F |
Novel |
Het |
|
N8 |
12.1 |
TPO
|
c.1810C > T |
p.R604W |
Novel |
Het |
|
N38 |
11.2 |
TSHR
|
c.1349G > A
|
p.R450H
|
Known
|
Het |
|
N9 |
56.2 |
DUOX2
|
c.2635G > A |
p.E879K |
Known |
Het |
|
N39 |
11.4 |
TSHR
|
c.1349G > A
|
p.R450H
|
Known
|
CoHet |
|
N10 |
29.2 |
TPO
|
c.2533G > A |
p.G845R |
Novel |
Het |
|
|
|
DUOX2
|
c.3383G > A |
p.R1128H |
Novel |
CoHet |
|
N11 |
14.7 |
DUOX2
|
c.2654G > T
|
p.R885L
|
Known
|
Het |
|
P1 |
27.6/0.5 |
DUOX2
|
c.4334T > A |
p.V1445E |
Novel |
ComHet |
|
N12 |
70.2 |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
ComHet |
|
|
|
DUOX2
|
c.3329G > A |
p.R1110Q |
Known |
|
|
|
DUOXA2
|
c.341G > C |
p.G114A |
Novel |
|
|
P2 |
21.7/0.89 |
DUOX2
|
c.3616G > A
|
p.A1206T
|
Known
|
ComHet |
|
N13 |
130 |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
Het |
|
|
|
DUOX2
|
c.3478delCTG |
p.L1160del |
Known |
|
|
N14 |
77 |
DUOXA2
|
c.280C > T |
p.R94C |
Novel |
Het |
|
P3 |
25.1 |
PAX8
|
c.177C > G |
p.S59R |
Novel |
Het |
|
N15 |
19.5 |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Het |
|
P4 |
21.2 |
DUOX2
|
c.4348T > C |
p.Y1450H |
Novel |
ComHet |
|
N16 |
10.1 |
DUOX2
|
c.3616G > A
|
p.A1206T
|
Known
|
Het |
|
|
|
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
|
|
N17 |
167 |
DUOXA2
|
c.535T > C |
p.Y179H |
Novel |
Het |
|
|
|
TSHR
|
c.1574T > C |
p.F525S |
Known |
Het |
|
N18 |
10.8 |
DUOX2
|
c.2654G > A
|
p.R885Q
|
Known
|
ComHet |
|
P5 |
23.6 |
PAX8
|
c.91C > T |
p.R31C |
Known |
Het |
|
|
DUOX2
|
c.2000delT |
p.L667fs |
Novel |
|
|
P6 |
1.3 |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Het |
|
N19 |
36 |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
Het |
|
P7 |
1.64 |
DUOX2
|
c.4171C > G |
p.P1391A |
Novel |
ComHet |
|
N20 |
11.2 |
DUOXA2
|
c.738C > G |
p.Y246X |
Known |
Het |
|
|
|
DUOX2
|
c.1946C > A
|
p.A649E
|
Known
|
|
|
N21 |
200 |
TPO
|
c.971T > C |
p.L324P |
Novel |
Het |
|
P8 |
11.5 |
DUOX2
|
c.2335G > A |
p.V779M |
Known |
ComHet |
|
N22 |
118 |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
Het |
|
|
|
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
|
|
N23 |
|
DUOX2
|
c.3239T > C
|
p.I1080T
|
Known
|
Het |
|
P9 |
0.03/0.89 |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Het |
|
N24 |
90.1 |
DUOX2
|
c.2654G > A
|
p.R885Q
|
Known
|
Het |
|
P10 |
23 |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Het |
|
N25 |
17.8 |
DUOX2
|
c.1883delA |
p.K628fs |
Known |
Het |
|
P11 |
19 |
TSHR
|
c.1349G > A |
p.R450H |
Known |
Het |
|
N26 |
41.6 |
DUOX2
|
c.2654G > A
|
p.R885Q
|
Known
|
Het |
|
P12 |
1.11/0.52 |
DUOX2
|
c.3239T > C
|
p.I1080T
|
Known
|
Het |
|
N27 |
13 |
TSHR
|
c.1349G > A
|
p.R450H
|
Known
|
Het |
|
P13 |
6.93/0.69 |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
Het |
|
N28 |
12 |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Het |
|
P14 |
12.2 |
DUOXA2
|
c.148C > T |
p.R50C |
Novel |
ComHet |
|
N29 |
10.3 |
TSHR
|
c.614T > C |
p.V205A |
Novel |
ComHet |
|
|
|
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
|
|
|
TSHR
|
c.1555C > T |
p.R519C |
Known |
|
|
P15 |
19.5/0.84 |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Het |
|
N30 |
16.4 |
DUOX2
|
c.1462G>A
|
p.G488R
|
Known
|
Het |
|
Table 3
In silico analysis of novel mutations
|
Gene |
AA change |
1000G*
|
ESP†
|
ExAC‡
|
KRG§
|
SIFT |
Polyphen2 |
LRT |
Mutation Taster |
Mutation Assessor |
FATHMM |
GERP |
|
TSHR
|
p.V205A |
NA |
NA |
NA |
NA |
D |
D |
D |
D |
M |
D |
6.02 |
|
PAX8
|
c.864+1G < T |
NA |
NA |
NA |
NA |
- |
- |
- |
D |
- |
- |
4.29 |
|
p.S59R |
NA |
NA |
NA |
NA |
D |
D |
D |
D |
H |
D |
5.31 |
|
TPO
|
p.R604W |
NA |
NA |
0.00002 |
NA |
D |
D |
N |
N |
L |
T |
4.24 |
|
p.G845R |
NA |
NA |
0.00004 |
NA |
T |
D |
- |
N |
N |
T |
- |
|
p.L324P |
NA |
NA |
NA |
NA |
D |
D |
D |
D |
H |
T |
4.99 |
|
DUOX2
|
p.E1469K |
NA |
0.0001 |
0.00006 |
NA |
D |
D |
D |
D |
M |
- |
5.68 |
|
p.L667fs |
NA |
NA |
NA |
NA |
- |
- |
- |
- |
- |
- |
- |
|
p.A1060V |
0.0009 |
NA |
0.00016 |
NA |
T |
B |
D |
D |
M |
- |
5.45 |
|
p.V407F |
NA |
NA |
NA |
NA |
D |
P |
D |
D |
M |
- |
2.14 |
|
p.R1128H |
NA |
NA |
0.00014 |
NA |
D |
D |
D |
D |
M |
- |
5.58 |
|
p.V1445E |
NA |
NA |
NA |
NA |
D |
D |
D |
D |
M |
- |
4.4 |
|
p.Y1450H |
NA |
NA |
0.00002 |
NA |
D |
D |
D |
D |
L |
- |
5.54 |
|
p.P1391A |
NA |
NA |
0.00001 |
NA |
D |
D |
D |
D |
H |
- |
5.84 |
|
DUOXA2
|
p.G114A |
NA |
NA |
NA |
NA |
- |
D |
D |
D |
L |
T |
4.66 |
|
p.Y179H |
NA |
NA |
0.00007 |
0.0016¶
|
D |
D |
D |
D |
M |
T |
5.3 |
|
p.R94C |
NA |
NA |
NA |
NA |
D |
D |
N |
N |
M |
T |
5.38 |
|
p.R50C |
NA |
NA |
NA |
NA |
D |
D |
D |
D |
M |
T |
5.77 |
Seven different mutations [p.G488R (n=13), p.A649E (n=3), p.R885Q (n=3), p.I1080T (n=2), and p.A1206T (n=2) in DUOX2; p.Y138X (n=9) in DUOXA2; and p.R450H (n=5) in TSHR) were recurrently observed as the mutations underlying CH. In addition, none of the mutations in SLC5A5 was found in our study population.
4. Genotype-phenotype relationship study
The relationship between TSH level and mutations showed no significant association under the following criteria: 1) allelic status (biallelic vs. monoallelic inactivation; P=0.1050), 2) classification of CH (genes associated with TD vs. genes associated with DH; P=0.2468), 3) mutated gene (TSHR vs. PAX8 vs. TPO vs. DUOX2 vs. DUOXA2; P=0.2582), and 4) study population (screening setting vs. clinic setting; P=0.3838).
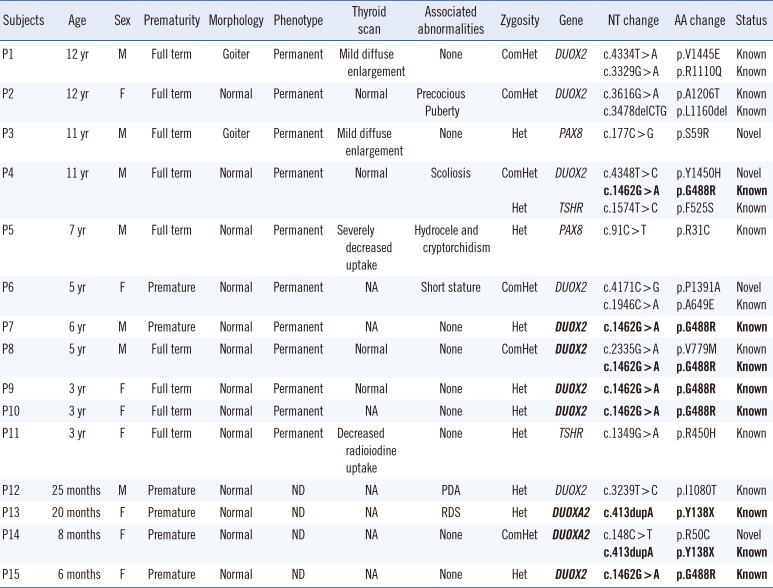
Among the 15 outpatients with mutations, four patients were preterm babies (
Table 4). Most of the patients with biallelic mutations in
DUOX2 (patient 1 [P1], P2, P4, P6, and P8) had permanent CH. Among them, manifestation of a goiter was found in only one patient (P1). The P4 with triallelic mutations (known heterozygous p.F525S in
TSHR and biallelic mutations in
DUOX2) showed no remarkable findings except permanent CH and scoliosis. The other two patients (P3 and P5) with monoallelic mutations in
PAX8 had permanent CH; 1 (P3 with p.S59R) with a history of cryptorchidism and hydrocele showed a mild diffuse enlargement of the thyroid gland, whereas the other (P5 with p.R31C) showed a marked decrease in the uptake of iodide on the thyroid scan. The P11 with a heterozygous mutation (p.R450H) in
TSHR showed a marked decrease in the uptake of iodide on the thyroid scan (
Table 4).
Table 4
Heterogeneous genotype-phenotype correlation in the Korean patients with congenital hypothyroidism
|
Subjects |
Age |
Sex |
Prematurity |
Morphology |
Phenotype |
Thyroid scan |
Associated abnormalities |
Zygosity |
Gene |
NT change |
AA change |
Status |
|
P1 |
12 yr |
M |
Full term |
Goiter |
Permanent |
Mild diffuse enlargement |
None |
ComHet |
DUOX2
|
c.4334T > A |
p.V1445E |
Known |
|
c.3329G > A |
p.R1110Q |
Known |
|
P2 |
12 yr |
F |
Full term |
Normal |
Permanent |
Normal |
Precocious Puberty |
ComHet |
DUOX2
|
c.3616G > A |
p.A1206T |
Known |
|
c.3478delCTG |
p.L1160del |
Known |
|
P3 |
11 yr |
M |
Full term |
Goiter |
Permanent |
Mild diffuse enlargement |
None |
Het |
PAX8
|
c.177C > G |
p.S59R |
Novel |
|
P4 |
11 yr |
M |
Full term |
Normal |
Permanent |
Normal |
Scoliosis |
ComHet |
DUOX2
|
c.4348T > C |
p.Y1450H |
Novel |
|
c.1462G > A
|
p.G488R
|
Known |
|
Het |
TSHR
|
c.1574T > C |
p.F525S |
Known |
|
P5 |
7 yr |
M |
Full term |
Normal |
Permanent |
Severely decreased uptake |
Hydrocele and cryptorchidism |
Het |
PAX8
|
c.91C > T |
p.R31C |
Known |
|
P6 |
5 yr |
F |
Premature |
Normal |
Permanent |
NA |
Short stature |
ComHet |
DUOX2
|
c.4171C>G |
p.P1391A |
Novel |
|
c.1946C>A |
p.A649E |
Known |
|
P7 |
6 yr |
M |
Premature |
Normal |
Permanent |
NA |
None |
Het |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
|
P8 |
5 yr |
F |
Full term |
Normal |
Permanent |
Normal |
None |
ComHet |
DUOX2
|
c.2335G > A |
p.V779M |
Known |
|
c.1462G > A
|
p.G488R
|
Known
|
|
P9 |
3 yr |
F |
Premature |
Normal |
Permanent |
Normal |
None |
Het |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
|
P10 |
3 yr |
M |
Premature |
Normal |
Permanent |
NA |
None |
Het |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
|
P11 |
3 yr |
F |
Premature |
Normal |
Permanent |
Decreased radioiodine uptake |
None |
Het |
TSHR
|
c.1349G > A |
p.R450H |
Known |
|
P12 |
25 months |
M |
Premature |
Normal |
ND |
NA |
PDA |
Het |
DUOX2
|
c.3239T > C |
p.I1080T |
Known |
|
P13 |
20 months |
F |
Premature |
Normal |
ND |
NA |
RDS |
Het |
DUOXA2
|
c.413dupA
|
p.Y138X
|
Known
|
|
P14 |
8 months |
F |
Premature |
Normal |
ND |
NA |
None |
ComHet |
DUOX2
|
c.148C > T |
p.R50C |
Novel |
|
c.413dupA
|
p.Y138X
|
Known
|
|
P15 |
6 months |
F |
Premature |
Normal |
ND |
NA |
None |
Het |
DUOX2
|
c.1462G > A
|
p.G488R
|
Known
|
Go to :
DISCUSSION
The current diagnostic tests for CH are based on morphological phenotype according to biochemical thyroid function tests, ultrasound, and thyroid scintigraphy. Although these tests can help pinpoint the underlying etiology, they generally provide incomplete information owing to phenotypic heterogeneity (the presence of the intermediate group, with neither TD nor DH). Knowing the exact underlying cause is not necessary during the newborn period because treatment starts without the etiology. However, a genetic diagnosis for CH has added benefits compared with the current phenotype-based approach. First, genetic screening for CH could allow the identification of mutation carriers at risk of recurrent subclinical hypothyroidism after the newborn period. Second, genetic screening might provide a definite etiologic diagnosis, thus providing insight into the molecular pathogenesis of CH. The genetic diagnosis might also be helpful in case where environmental factors may modify the phenotype of CH, leading to false-negative results.
Thyroid dysfunction detected on NBS is highly heterogeneous; it results from a variety of causes, such as true CH, hyperthyrotropinemia, and subclinical hypothyroidism. A recent study revealed that approximately 30% of newborns with false-positive results in NBS developed persistent subclinical hypothyroidism in their late childhood periods, probably due to genetic abnormalities [
23]. Here, we conducted genetic screening for six genes in cases with thyroid dysfunction detected on NBS. This study revealed that there were genetic abnormalities underlying CH in 35% (39/112) of the newborns and in 24% (14/58) of the outpatients. The proportion of mutations was considerably higher than expected in the Korean newborn population; it is known that gene mutations are found in only 2% of cases with TD, which account for 85% of CH [
24]. This study points to an underestimation of the genetic component in the previous studies, in which genetic screening was conducted without consideration of the intermediate phenotypes [
1125]. We speculate that there might be yet undetermined genetic components of thyroid dysfunction in NBS.
The genetic incidence based on the mutations of six genes was estimated to be 1 in 6,580 in the Korean newborns.
DUOX2 mutations were a frequent cause of CH associated with mutations in both the NBS setting (17%, 19/112) and outpatient setting (16%, 9/58). A recent study regarding Korean patients reported that
DUOX2 mutations are a common cause of CH (35%, 15/43) [
12]. It should be noted that the inclusion criteria for outpatients in our study (primary CH) differed from that in the previous study (CH with eutopic thyroid glands), thus explaining the higher proportion of
DUOX2 mutations in the previous study [
12]. Inone Chinese study, biallelic mutations in
DUOX2 have been identified in approximately 15% (10/67) of patients with CH and goiters [
14]. Another Japanese study reported biallelic mutations in
DUOX2 in 28% (4/14) of the patients with permanent DH and 4% (4/102) of the patients with permanent CH. In the present study, biallelic mutations in
DUOX2 were identified in 9% of the patients with primary CH. Considering that most of our outpatients had permanent CH, the contribution of
DUOX2 mutations in the Korean population were higher than that in the Japanese. In contrast,
TPO mutations were not a major cause of CH in our study, although they have been reported as the most prevalent cause of DH in Caucasians [
8].
In the current study, there was a trend that biallelic inactivation was more frequently observed in outpatients than in newborns, even though there was no statistical significance (P=0.059). The mutation spectrum between them was quite similar, as recurrent mutations in DUOX2, DUOXA2, and TSHR were observed in both newborns and outpatients. Furthermore, we revealed that the genetic screening of outpatients (14/58, 24%) might lead to an underestimation of mutations compared with the genetic screening of newborns (39/112, 35%). Considering the lower positive rate of mutations in outpatients, there might be an incomplete penetrance of mutations for clinical manifestations of CH.
As the previous studies noted, phenotypic manifestations in our outpatients were observed in cases with both monoallelic and biallelic mutations [
714171819]. One reason for the high proportion of heterozygote mutations might be attributed to false negatives, unidentified mutations in intronic and regulatory regions, allele dropout due to single-nucleotide polymorphisms in PCR primer binding sites, or structural variations, such as large deletions. Apart from the possibility of false negatives, monoallelic mutations could be sufficient for the pathogenesis of CH.
Recently, the possibility of a non-Mendelian mechanism of CH has been raised, and oligogenic inheritance, triallelic mutations, or synergistic heterozygosity have been suggested [
1520]. In this study, there was one outpatient with permanent CH associated with triallelic mutations. The patient's phenotype owing to the biallelic
DUOX2 mutations was not aggravated by the coexisting monoallelic
TSHR mutation. Recently, it has been reported that triallelic mutations in
DUOX2 were associated with permanent CH [
20]. Genotype-phenotype correlation studies showed that biallelic/monoallelic mutations in
DUOX2 are associated with permanent CH, whereas monoallelic mutation in
TSHR has been reported in mild CH [
714]. However, little is known about the combined effects of the triallelic mutations involving the two genes. Triallelic mutations involving two different genes have also been reported in other diseases such as telomeropathies [
26]. The presence of triallelic mutations is consistent with the concept of synergistic heterozygosity, meaning that multiple heterozygotes in metabolic pathway genes could lead to the disease phenotype [
27]. Previous studies on CH reported coexistence of
DUOX2/
TSHR or
TPO/
TSHR mutations in the same individuals [
1215]. The relationship between the coexisting mutant alleles and the phenotypic manifestations was not conclusive in the previous studies [
1215]. In the present study, we could not determine the functional and phenotypic effects of triallelic mutations. Although synergistic heterozygosity would be a good explanation, experimental studies and more genotype-phenotype correlation studies are needed to determine whether the phenotype was influenced by the combination of the mutant alleles. Taken together, the genotype-phenotype correlation is highly complex in CH.
One of the important limitations of this study is that we could not perform functional experiments, although comprehensive in silico analyses were done. Another limitation is that we could not completely evaluate genotype-phenotype correlations in newborns with thyroid dysfunction. Furthermore, we did not cover other genes for CH such as TG or DEHAL1.
In summary, despite the noted limitations, we determined the mutation incidence of CH in a routine NBS setting through population-based genetic screening. This study showed that genetic screening that excludes the intermediate group (neither TD nor DH) might lead to an underestimation of mutations. In addition, we concluded that DUOX2 mutations were a frequent cause of CH in the Korean population as well as in the Korean patients.
Go to :


 PDF
PDF ePub
ePub Citation
Citation Print
Print


 XML Download
XML Download