

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Cornelia de Lange Syndrome (CdLS) is a rare, clinically heterogeneous congenital anomaly presenting with distinctive facial features, growth retardation, hirsutism, and upper limb reduction [12]. Although trained clinicians easily recognize classical cases of CdLS, about 20-30% of the patients exhibit only mild phenotypes [3]. The rarity and highly varied presentation of CdLS has limited the ability to make an accurate diagnosis based on clinical diagnostic criteria alone [4]. Thus, additional diagnostic procedures are required to improve the accuracy of CdLS diagnosis.
In addition to the variable phenotypes of CdLS, its genetic heterogeneity creates significant challenges for both diagnosis and genetic counseling [56]. Currently, five genes are known to be associated with CdLS: NIPBL, SMC1A, SMC3, RAD21, and HDAC8, which are all regulators or structural components of sister chromatid cohesion [6]. NIPBL, SMC3, and RAD21-related CdLS are inherited in an autosomal dominant manner, while SMC1A and HDAC8-related CdLS are inherited in an X-linked manner. A half of the affected individuals have an NIPBL mutation [7], and about 10% of the patients have mutations in the other four genes [6]. Herein, we describe a family with CdLS carrying a novel pathogenic variant of the SMC1A gene that was identified by exome sequencing.
The proband (III:1, Fig. 1A) was a 3-yr-old boy presenting with a developmental delay. The proband was delivered preterm spontaneously at 35 weeks. His facial dysmorphism was noted: arched, bushy eyebrows extending down onto the nasal bridge, low hairline, broad nasal bridge with anteverted nares, low-set and outwardly placed ears, long philtrum, thin upper lip, and hirsutism. His hands were small but not malformed. Bilateral clinodactyly of the fifth finger was also evident. The patient exhibited motor and language delays: he began to walk independently at 24 months of age and could say only one word at three years of age. On physical examination at three years of age, his height and weight were below the 3rd percentile.
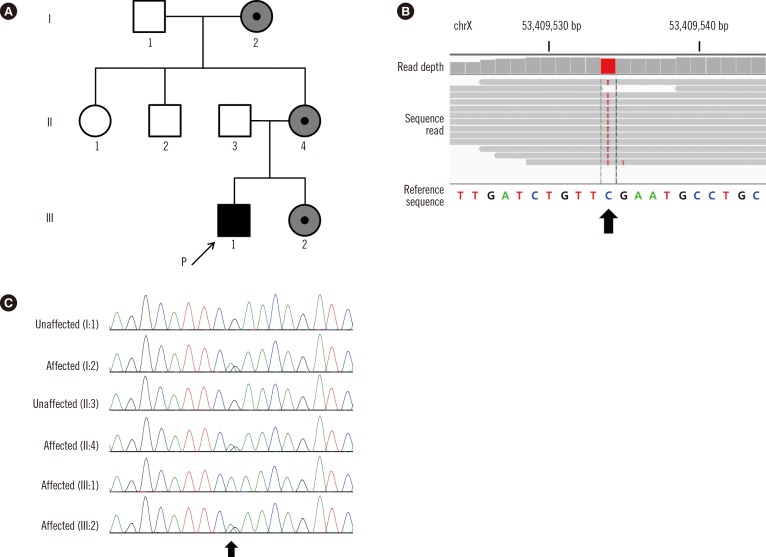 | Fig. 1Pathogenic SMC1A variant in a CdLS family. (A) Pedigree of the family, with four related cases of CdLS. The arrow indicates the index patient. A black or gray symbol indicates clinically affected family members (gray, mildly affected). (B) Integrative Genomics Viewer snapshot of the novel SMC1A pathogenic variant (NM_006306.3:c.3178G>A, p.Glu1060Lys) identified by exome sequencing (arrow). (C) Sequence analysis of the SMC1A gene. Chromatograms show the hemizygous nonsynonymous variant (c.3178G>A; p.Glu1060Lys) of the SMC1A gene in the proband (III:1), the heterozygous variant in individuals I:2, II:4, and III:2 (II:4 and III:2 are very mildly affected), and the normal sequence in unaffected subjects I:1 and II:3 (arrow).
|
The proband's younger sister (III:2) showed delayed verbal development with mild mental retardation (IQ of 67). The proband's mother (II:4) and grandmother (I:2) are of short stature (less than the 5th percentile). They had learning disabilities and showed a slight impairment in cognitive development. Dysmorphic facial features were not remarkable, and hearing loss was not observed in this family.
Based on the clinical signs, CdLS was suspected. However, no NIPBL mutation was identified by Sanger sequencing. Subsequently, we decided to perform exome sequencing for the following reasons: (1) CdLS-like disorders associated with developmental delay or facial dysmorphism could not be excluded, (2) conventional gene-by-gene sequencing is too expensive and time-consuming, and (3) exome sequencing allows simultaneous analysis of all CdLS candidate genes.
After obtaining informed consent, genomic DNA was extracted and captured with Agilent SureSelect Human All Exon v3 Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a HiSeq2000 platform (Illumina, Inc., San Diego, CA, USA). After screening all CdLS-related genes, we identified a missense variant (NM_006306.3:c.3178G>A, p.Glu1060Lys) in the SMC1A gene (Fig. 1B). The p.Glu1060Lys variant was absent from dbSNP (build 135) and the Exome Sequencing Project database (http://evs.gs.washington.edu/EVS/) and was not detected in our in-house variant database consisting of 96 Korean exomes of various inherited disorders other than CdLS. Bioinformatic analysis revealed that the affected residue, Glu1060, is strictly conserved from zebrafish to humans, and both SIFT (http://sift.bii.a-star.edu.sg/index.html) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) predicted p.Glu1060Lys to be deleterious. Thus, we concluded that the identified variant was most likely the causative mutation.
Sanger sequencing of the SMC1A exon 21 was performed. Proband (III:1) was hemizygous for the variant (c.3178G>A; p.Glu1060Lys) in the SMC1A gene (Fig. 1C). The family study showed that his sibling (III:2), mother (II:4), and grandmother (I:2) were heterozygous for the variant.
The SMC1A gene is located on chromosome Xp11.22 and consists of 26 exons spanning nearly 48.6 kb in total. Many variants of the SMC1A gene, mostly missense mutations, are spread throughout the coding region. Although several Korean CdLS cases with NIPBL mutations have been reported [89], no cases with SMC1A mutations have been noted to date.
In 2006, Musio et al. [10] identified SMC1A as a causative gene for CdLS. Deardorff et al. [3] reported that many allelic variants of SMC1A, including SMC3, contribute to approximately 5% of all cases of CdLS. The phenotypes of the affected individuals reported in these studies indicate that mutations in SMC1A result in milder forms of CdLS, with no predominant structural anomalies, but with notable cognitive involvement [310]. To address the genotype-phenotype correlation in Korean patients with CdLS, we compared the clinical features of the family with the SMC1A pathogenic variant from this study and those of the three previously reported patients with NIPBL mutations (Table 1). The phenotype of our patient included typical facial features of CdLS, but no limb or digit reduction or other major structural anomalies.
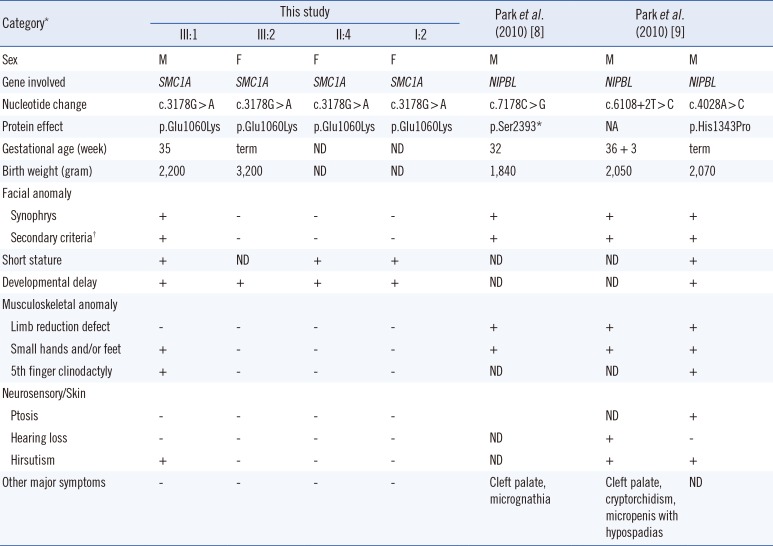
Table 1
Genotype-phenotype correlation analysis of Korean CdLS patients harboring SMC1A or NIPBL mutations
| Category* | This study | Park et al. (2010) [8] | Park et al. (2010) [9] | ||||
|---|---|---|---|---|---|---|---|
| III:1 | III:2 | II:4 | I:2 | ||||
| Sex | M | F | F | F | M | M | M |
| Gene involved | SMC1A | SMC1A | SMC1A | SMC1A | NIPBL | NIPBL | NIPBL |
| Nucleotide change | c.3178G > A | c.3178G > A | c.3178G > A | c.3178G > A | c.7178C > G | c.6108+2T > C | c.4028A > C |
| Protein effect | p.Glu1060Lys | p.Glu1060Lys | p.Glu1060Lys | p.Glu1060Lys | p.Ser2393* | NA | p.His1343Pro |
| Gestational age (week) | 35 | term | ND | ND | 32 | 36 + 3 | term |
| Birth weight (gram) | 2,200 | 3,200 | ND | ND | 1,840 | 2,050 | 2,070 |
| Facial anomaly | |||||||
| Synophrys | + | - | - | - | + | + | + |
| Secondary criteria† | + | - | - | - | + | + | + |
| Short stature | + | ND | + | + | ND | ND | + |
| Developmental delay | + | + | + | + | ND | ND | + |
| Musculoskeletal anomaly | |||||||
| Limb reduction defect | - | - | - | - | + | + | + |
| Small hands and/or feet | + | - | - | - | + | + | + |
| 5th finger clinodactyly | + | - | - | - | ND | ND | + |
| Neurosensory/Skin | |||||||
| Ptosis | - | - | - | - | ND | + | |
| Hearing loss | - | - | - | - | ND | + | - |
| Hirsutism | + | - | - | - | ND | + | + |
| Other major symptoms | - | - | - | - | Cleft palate, micrognathia | Cleft palate, cryptorchidism, micropenis with hypospadias | ND |
The reference sequences used are NM_006306.3 (SMC1A) and NM_133433.3 (NIPBL).
*Diagnostic criteria for CdLS reported in the literature [4]; †Long eyelashes, short nose with anteverted nares, long philtrum, broad or depressed nasal bridge, small or square chin, thin lips, high palate, and widely spaced or absent teeth.
Abbreviations: M, male; F, female; NA, not applicable; ND, no data.
![]()
The SMC1A gene encodes the SMC1A protein that is one of the three core cohesion subunits (SMC1, SMC3, and RAD21) [1]. The SMC protein, which is the cohesion complex, is involved in the structural maintenance of chromosome with the essential role for the proper segregation of sister chromatids during cell division [11]. It also plays a fundamental role in DNA-damage repair and regulation of gene expression [11]. The SMC proteins contain N- and C-terminal ATP-binding domains, and two extended coiled-coil domains separated by a hinge domain [12]. The p.Glu1060Lys variant identified in our study is located in the highly conserved coiled-coil domain of the SMC1A protein.
In conclusion, we identified a novel pathogenic variant (c.3178G>A; p.Glu1060Lys) of the SMC1A gene causing a mild phenotype of CdLS. Our results reinforce the consensus that SMC1A gene defects are associated with a milder phenotype of CdLS. Furthermore, exome sequencing can be a useful tool to identify causative pathogenic variants in patients with CdLS.
 XML Download
XML Download