

 PDF
PDF ePub
ePub Citation
Citation Print
Print


21-Hydroxylase deficiency is the most common cause of congenital adrenal hyperplasia (CAH, OMIM 201910). More than 90% of patients with CAH have CYP21A2 mutations including conversions to the CYP21A1P pseudogene or large deletions [1]. 21-Hydroxylase-deficient CAH is an autosomal recessive disorder that is manifested in a variety of clinical severities comprised of three subtypes: (1) classic salt-wasting, (2) classic simple virilizing, and (3) non-classic forms [2]. The incidence of classic CAH ranges from 1 in 10,000 to 1 in 20,000 births worldwide [2].
Analysis of the CYP21A2 mutation is challenging owing to the presence of a highly homologous CYP21A1P pseudogene (98% in exons and 96% in introns) [3], which is known to interfere with targeted CYP21A2 amplification during sequencing. Furthermore, the RCCX module (the genes RP, C4, CYP21, and TNX arranged in tandem) in the 6p21.33 chromosome region shows high homology between the functional genes (RP1, CYP21A2, and TNXB) and the corresponding pseudogenes (RP2, CYP21A1P, and TNXA). This phenomenon leads to gene conversions and gene deletions due to homologous recombination, which results in inactivation of the functional gene [4]. Since a large number of CYP21A2 mutations are composed of large pseudogene conversions and deletions (20-30%), additional modalities other than direct sequencing are essential to accurately identify these mutations. The objective of this study was to accurately analyze the CYP21A2 genotype using a combination of complementary methods.
Genetic analyses were performed in retrospectively selected 14 patients (eight males and six females) with CAH who were referred for CYP21A2 mutation analysis from 2008 to 2013 at Samsung Medical Center, Seoul, Korea. Six family members from three of these patients were also included in the study. Informed consent was obtained from the patients or the parents for pediatric patients. The Institutional Review Board of Samsung Medical Center approved this study.
All CAH-suspected patients showed elevated levels of 17-hydroxyprogesterone (17-OHP). Nine were classified as salt-wasting, two were non-classic, and one patient was simple virilizing according to clinical features. One female patient only presented with irregular menstruation (case 8), and clinical information was not available for one patient (case 4).
Genomic DNA was extracted using Wizard Genomic DNA Purification kits (Promega, Madison, WI, USA) according to the manufacturer's instructions. We performed long-range PCR using the AccuPower Hotstart PCR PreMix (Bioneer, Daejeon, Korea), containing a high-fidelity polymerase, buffers, and a deoxynucleotide triphosphate mix. The reaction mixture included 500 ng of DNA and 10 pmol each of CYP779f and Tena32F primers [5]. The PCR amplification conditions were: 94℃ for 5 min followed by 32 cycles at 94℃ for 30 sec, 60℃ for 30 sec, 72℃ for 1 min, and a final extension at 72℃ for 7 min. The resultant product of CYP779f and Tena32F primers and the subsequent TaqI restriction endonuclease-digested products were analyzed by electrophoresis in 1.2% agarose gels as previously described [67]. The presence of an 8.5-kb PCR product and the appearance of TaqI-produced 3.7- and 2.5-kb fragments represent intact CYP21A2 and partial fragments of the TNXB gene in normal individuals, whereas TaqI-produced fragments that are 3.2 and 2.4 kb in size represent CYP21A1P pseudogene recombinations in patients with CAH [7]. An aliquot of the same CYP779f and Tena32F PCR product was subsequently submitted for direct sequencing of all the coding exons and the flanking intronic sequences for CYP21A2, using primers designed in this study (available on request). Direct sequencing was performed by using an ABI Prism 3130xl Genetic Analyzer (Life Technologies, Carlsbad, CA, USA), and sequence variants were designated according to Human Genome Variation Society recommendations (http://www.hgvs.org/mutnomen/) by using the reference sequences from GenBank (NG_007941.2, NM_000500.7, NP_000491.3).
The multiple ligation-dependent probe amplification (MLPA) assay was performed concurrently by using the SALSA MLPA kit P050-B2 (MRC-Holland, Amsterdam, Netherlands) according to the manufacturer's instructions. The kit is designed to detect large deletions and duplications in one or more exons of the CYP21A and TNX genes, and contains five probes for different CYP21A2 mutations and three CYP21A1P-specific probes. The MLPA data were analyzed by using GeneMarker v.1.9 software (SoftGenetics, State College, PA, USA).
We identified 27 mutations in 28 alleles of the 14 CAH-suspected patients. The clinical features and molecular data of the patients and family members are listed in Table 1. Eight different point mutations [c.92C>T (p.Pro31Leu), c.293-13A>G or c.293-13C>G, c.518T>A (p.Ile173Asn), c.844G>T (p.Val 282Leu), c.874G>A (p.Gly292Ser), c.955C>T (p.Gln319*), c.1054G>A (p.Glu352Lys), c.1069C>T (p.Arg357Trp)], three small deletions and/or insertions [c.923dupT (p.Leu308 Phefs*6), c.1451_1452delGGinsC (p.Arg484Profs*58), c.492 delA (p.Glu164Aspfs*24)], and one mutation cluster were identified. The mutation cluster was on exon 6, and it was defined as c.[710T>A;713T>A;719T>A] (p.[Ile237Asn;Val238Glu;Met240Lys]). The most common mutation was the intron 2 splice site mutation (c.293-13A>G or c.293-13C>G) at 25%, followed by the p.Ile173Asn mutation with an allele frequency of 17.9%. Three or more mutations were detected in three patients (patients 7, 9, and 14).
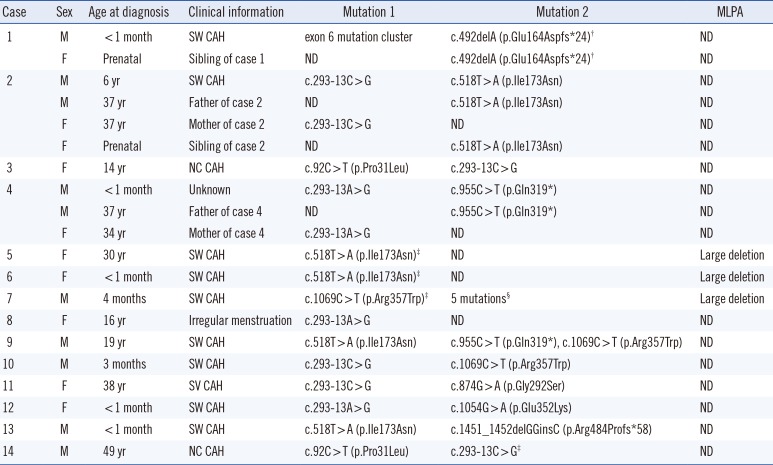
Table 1
Clinical and genetic characteristics of 14 CAH-suspected cases and six family members
†Novel mutation; ‡Homozygous pattern in sequencing analysis chromatograms; §5 mutations: c.92C>T (p.Pro31Leu), exon 6 mutation cluster, c.844G>T (p.Val282Leu), c.923dupT (p.Leu308Phefs*6), c.955C>T (p.Gln319*).
Abbreviations: CAH, congenital adrenal hyperplasia; SW, salt-wasting type; NC, non-classic type; SV, simple virilizing; ND, not detected; exon 6 mutation cluster, c.[710T>A; 713T>A; 719T>A] (p.[Ile237Asn; Val238Glu; Met240Lys]); MLPA, multiple ligation-dependent probe amplification.
![]()
Approximately 95% of the defective CYP21A2 mutations seen in CAH patients fall into three categories [4]: (i) approximately 61 to 70% [891011121314] are deleterious mutations due to small gene conversions derived from the CYP21A1P pseudogene, including c.293-13A>G or c.293-13C>G (20.6-30.3%), p.Ile173Asn (8.2-19.8%), p.Val282Leu (2.2-26.2%), p.Arg357Trp (3.0-8.4%), p.Gln319* (2.4-6.7%), p.Gly111Valfs*21 (0.8-4.3%), exon 6 mutation cluster [p.Ile237Asn, p.Val238Glu, p.Met240Lys (1.1-3.0%)], and p.Pro31Leu (0.3-2.6%); (ii) approximately 5% of defective CYP21A2 mutations are relatively infrequent spontaneous mutations; and (iii) approximately 7.5 [10] to 32.2% [11] are large gene rearrangements generated by unequal meiotic crossover. In our study, six different mutations derived from the CYP21A1P pseudogene due to small gene conversions (p.Pro31Leu, c.293-13A>G or c.293-13C>G, p.Ile 173Asn, exon 6 mutation cluster, p.Gln319*, and p.Arg357Trp) were detected in 20 alleles (71.4%). Three relatively infrequent mutations (p.Gly292Ser, p.Glu352Lys, and p.Arg484Profs*58) were detected in three alleles (10.7%), and three large deletions were found in three alleles (10.7%). These findings implicate that although we performed molecular genetic analysis of CYP21A2 in a limited number of cases, our mutational spectrum corresponded with previous data generated from large cohorts [891011121314].
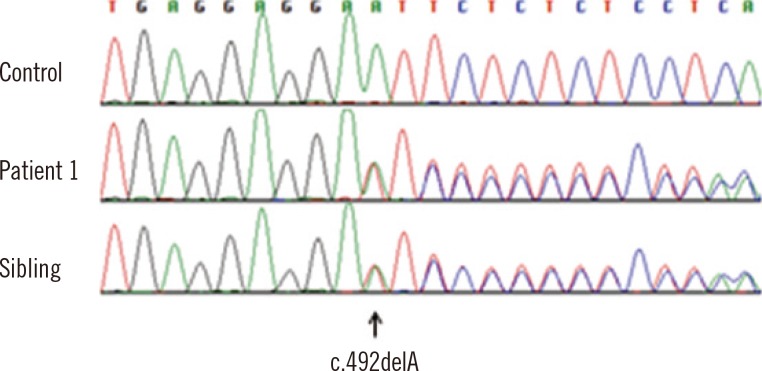
Patient 1 had a novel frame-shift mutation, c.492delA (p.Glu164Aspfs*24) (Fig. 1), and an exon 6 mutation cluster. Clinically, the patient showed markedly elevated 17-OHP in neonatal screening with salt-wasting signs and was treated with fludrocortisone and hydrocortisone. The sibling of patient 1 underwent prenatal mutation analysis with cultured chorionic villi samples and had only one mutant allele, c.492delA.
Patient 8, who had only one heterozygous mutation (c.293-13A>G), was a 16-yr-old female who visited the obstetrics outpatient clinic for evaluation of irregular menstruation for three years without virilization. She showed an elevated basal 17-OHP level (620 ng/dL, reference range: <200 ng/dL) without abnormal results for testosterone or estradiol, which could imply non-classic type CAH according to the guidelines on CAH by Speiser et al. [2]. To rule out non-classic type CAH, she was evaluated after ACTH stimulation. The patient's 17-OHP level at 60 min after ACTH stimulation was 839 ng/dL, which was compatible with a genetically heterozygous carrier status. This patient is being followed up without any treatment.
Large deletions of CYP21A2 were detected in three alleles (10.7%) by MLPA analysis. Of them, patients 5 and 7 showed compatible results for heterozygous deletion of entire CYP21A2 (exons 1 to 8), and patient 6 showed results implying partial heterozygous deletion from exons 1 to 6. New et al. [8] reported the frequency of deletion and large gene conversions at 20.0% in the largest tested cohort to date including 1,507 families with heterogeneous ethnic backgrounds. Other relatively large studies that included different ethnicities showed variable deletion and large gene conversion frequencies from 11.2-32.2% [9111213]. Ma et al. [14] showed rates of 15% for large deletions and 15% for conversions from 30 Chinese patients. However, Lee et al. [10] presented a lower occurrence of large gene deletions (or conversions) at 7.5% of the CYP21A2 alleles in ethnic Chinese (Taiwanese) patients. Previous Korean studies [151617] had used different methods from those of the current study to analyze CYP21A2 mutations, and the largest study by Choi et al. [17] determined the frequency of large deletions to be 31.3%. The differences in the frequency of large deletions between previous reports [891011121314151617] and our data may have resulted from differences in population size between the studies and/or differences in mutation analysis methods.
In terms of genotype-phenotype correlations, nine salt-wasting-type CAH patients had one or more mutations known to be associated with more severe CAH phenotypes (exon 6 mutation cluster, c.293-13A>G or c.293-13C>G, p.Ile173Asn, p.Gln319*, p.Arg357Trp, p.Arg484Profs*58, and large deletion) [8], while two patients with non-classic type CAH had the p.Pro31Leu mutation, which is frequently associated with non-classic CAH.
In a recent report by Xu et al. [18], a molecular analysis strategy for examining CYP21A2 in CAH patients was discussed. They proposed a sequential approach according to primary testing using a PCR-restriction fragment length polymorphism (PCR-RFLP) method with two different sets of primers and MLPA. Then, patients with or without deletions proceeded to sequencing of the CYP779f/Tena32F amplicon, and patients with duplications proceeded to sequencing with an amplicon using an additional set of primers [18]. Although duplications are rare (1.53%) and were not detected by MLPA in our study, this possible limitation of the current method should not be overlooked when duplications are detected.
In the present study, we have employed complementary methods to analyze CYP21A2. The presence of pseudogene and a high incidence of gene conversion and large deletions of the CYP21A2 could lead to analytic failure or post-analytic misinterpretation. Without complementary methods, when a homozygous mutant peak is detected, a hemizygous mutation due to a large deletion might be misinterpreted as a homozygous mutation in the absence of gene dosage analysis. The use of long range PCR and RFLP analysis confirmed valid CYP21A2 amplification and also large scale deletion and/or conversion events before direct sequencing. In addition, MLPA assays were conducted for gene dosage analysis along with determination of approximate location and range of dosage mutations. In conclusion, we successfully identified CYP21A2 mutations in CAH-suspected patients using both long-range PCR and RFLP analyses followed by sequencing and concurrent MLPA analysis.
 XML Download
XML Download