

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Bardet-Biedl syndrome (BBS) (OMIM 209900) is an autosomal recessive, clinically and genetically heterogeneous ciliopathy [1, 2]. The prevalence of BBS is relatively low, ranging from 1:100,000 in the North American to 1:160,000 in the European population, and is rarely found in East Asia [3, 4]. To date, at least fifteen BBS genes (BBS1-14, SDCCAG8) have been identified, accounting for 70-76% of BBS cases. Among them, BBS 1, 2, 10, and 12 are considered the major causative genes [3, 4, 5]. Correlation between the BBS genotype and phenotype varies among and within families [1, 3]. Here, we report the first genetically confirmed BBS case in a Korean family with a compound heterozygous mutation of the BBS7 gene.
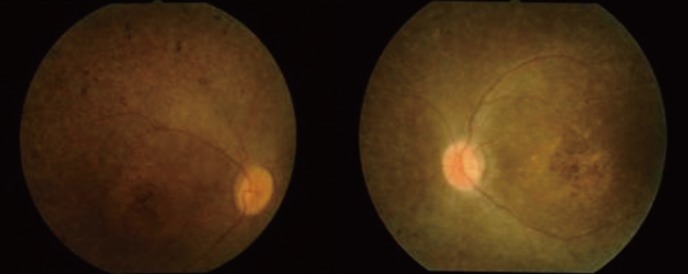
A 26-yr-old Korean male (proband) was the second son of non-consanguineous Korean parents. He was blind, mentally retarded, and truncally obese. Past history showed that he had undergone an operation for an atrial septal defect at the age of four. A chest X-ray revealed cardiomegaly and pulmonary edema. Renal sonogram revealed bilateral small-sized kidneys, increased renal parenchymal echogenicity, and poor corticomedullary differentiation, together with laboratory findings, were indicative of an end-stage renal disease. Fundus examination revealed pigmentary changes as typically observed in retinitis pigmentosa with optic disc atrophy (Fig. 1). The proband was treated with maintenance hemodialysis and antihypertensive medications.
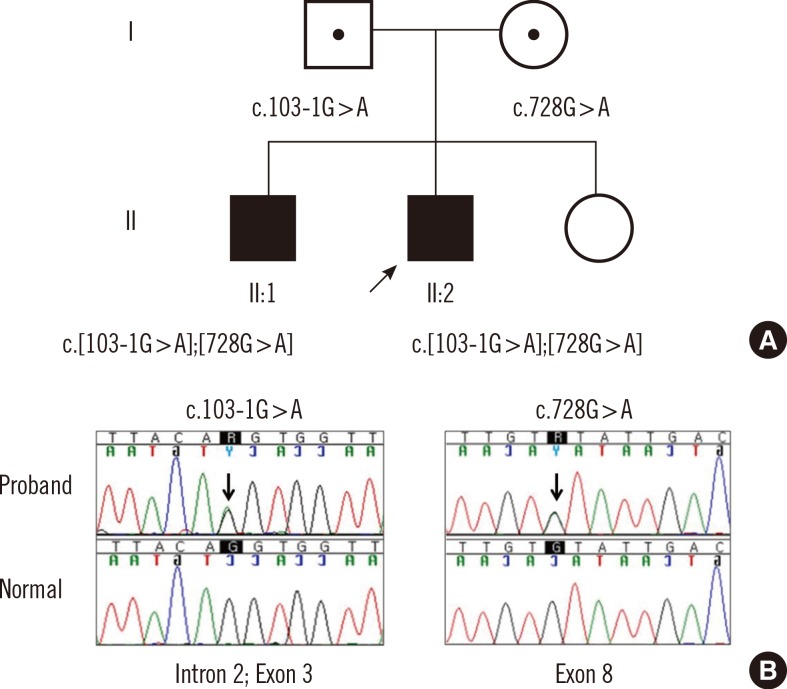
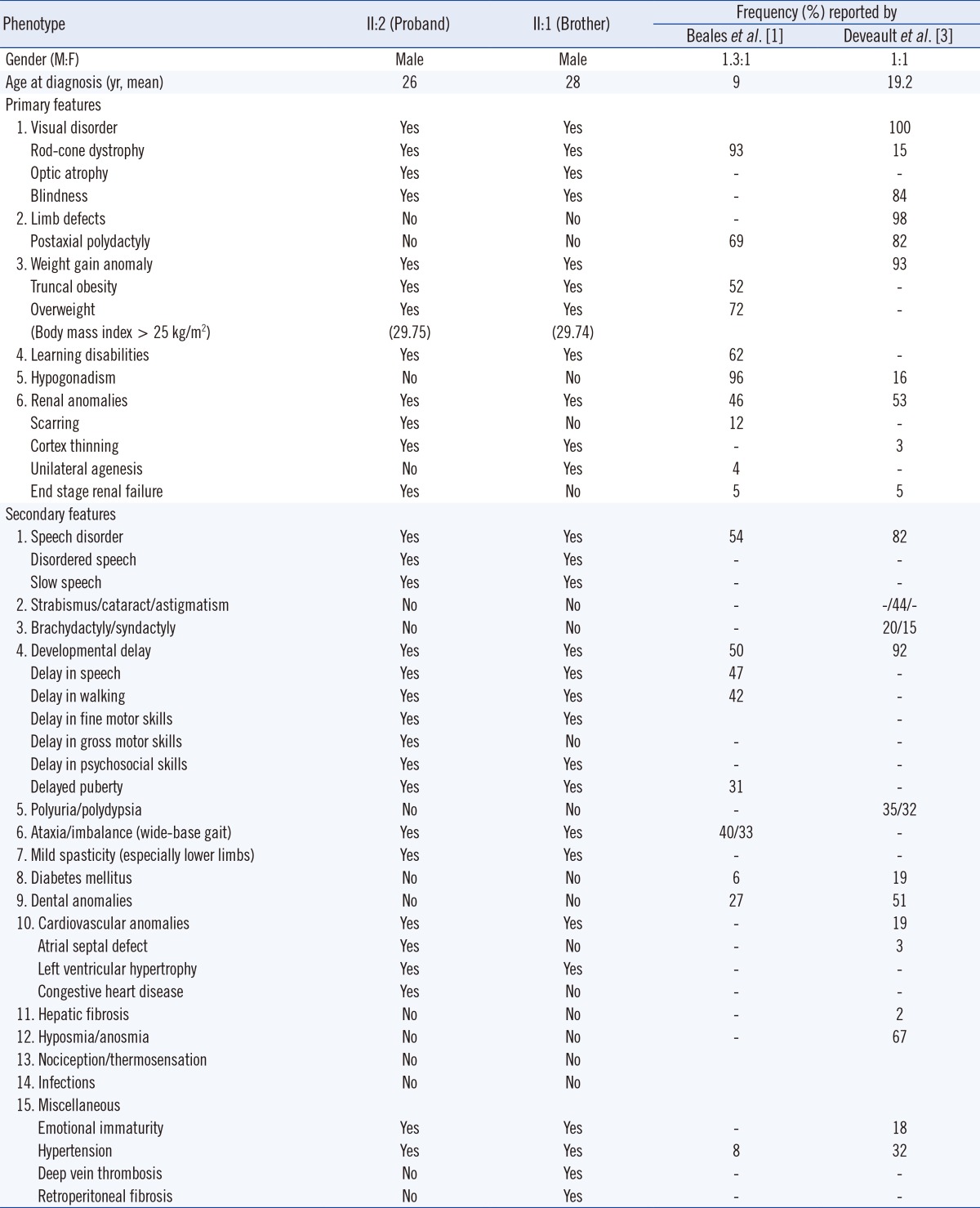
The proband's 28-yr-old brother showed similar clinical findings, but his parents, younger sister, and other blood relatives presented as non-specific (Fig. 2A). Clinical characteristics of the proband and his brother are summarized in Table 1, and both of their diagnoses were consistent with the BBS diagnostic criteria [1, 2]. However, his brother showed some different phenotypes: unilateral kidney agenesis with milder renal symptoms, no atrial septal defect with milder cardiovascular symptoms, and deep vein thrombosis of the left lower extremity.
Genetic studies conducted on the fourteen known BBS genes (BBS1-BBS14) from all family members by Sanger sequencing revealed that the proband and his brother shared the same compound heterozygous variants of the BBS7 gene (NM_176824.2) (Fig. 2B). One of which was a novel variant (c.103-1G>A) in the consensus splice acceptor site, which altered the splicing recognition site of 'AG' to 'AA' at the BBS7 gene intron 2 and exon 3 boundary. In silico analysis of the mutant splice site using a neural network splice site scoring program (NNSPLICE 0.9 version) predicted that the splice site would be abolished. The other variant was a c.728G>A (p.Cys243Tyr) missense mutation, which has been reported in a case describing juvenile retinitis pigmentosa, by Wang et al. [6]. The father was a heterozygous carrier for c.103-1G>A, and the mother was a heterozygous carrier for c.728G>A. Additionally, array comparative genomic hybridization analysis using the NimbleGen 3×720 K array (Roche NimbleGen Inc., Madison, WI, USA) revealed a normal hybridization pattern with no evidence of any significant chromosomal imbalance.
BBS is a ciliopathy involving multiple systems, characterized by a primarily autosomal recessive inheritance, but three mutational alleles in two BBS genes have been implicated (trialleic inheritance) in some families [4, 7]. No additional pathogenic allele was found in other BBS genes, from BBS1 to BBS14, in this case. BBS proteins are known to play an important role in ciliary biogenesis and signaling [2, 8], and BBS7 protein dysfunctions or abnormalities can cause structural and functional defects in cilia, and as a result, affect variable organs in the body [9, 10].
The present case showed neither polydactyly, as usually seen in 69-82% of affected individuals, nor hypogonadism, usually seen in 16-96% of affected individuals according to previous reports (Table 1) [1, 3]. Interestingly, minor phenotypic variabilities, including renal and cardiac anomalies were also identified within the family. Therefore, further work is required to differentiate BBS from the other ciliopathies, because the spectrums of BBS can overlap with the other ciliopathies such as the McKusick-Kaufan syndrome, which involves the limb, cardiac and associated urogenital systems, with BBS6 as the causative gene; or the Alström syndrome, which leads to obesity, retinal and endocrine systems with ALMS1 as the causative gene [1, 2, 3].
Nevertheless, 24-30% of BBS cases show no mutations in BBS genes [3, 4, 5]. BBS phenotype could be affected not only by transcription of DNA mutations, but also by other etiologies not addressed in this study, such as protein defects associated with noncoding RNA regulatory mechanisms or methylation. Further studies are warranted to clarify these issues.
 XML Download
XML Download