

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Stem cell transplantation (SCT) has become an important treatment for hematological malignant disorders. However, SCT may cause severe complications, including graft rejection, relapse of malignancy, veno-occlusive disease, infections, and graft-versus-host disease. To maximize compatibility and minimize complications of allogeneic hematopoietic SCT, selection and evaluation of the best possible donor are crucial. This assessment should include HLA typing, complete medical history, chest X-ray, electrocardiogram, blood screening, and infectious disease screening [1, 2].
Donor chromosome analysis remains controversial and is not performed in many institutions when evaluating donors. However, there are rare reports on the transmission of constitutional karyotype abnormalities after SCT [3]. Here, we report the identification of a constitutional t(2;11)(q32;q23) chromosomal abnormality in a pediatric patient with acute megakaryoblastic leukemia (AMKL) and a sibling donor after allogeneic bone marrow transplantation (BMT).
A 10-month-old male was admitted for clinical evaluation of leukocytosis. He presented with irritability, fever, and marked leukocytosis (46.41×109/L). On his bone marrow (BM) aspirate smear, 68% of the blasts showed irregular cytoplasmic blebs. The blasts were negative for myeloperoxidase, Sudan-Black B, and periodic acid-Schiff staining. According to the flow cytometry that was conducted on the guardian consent, the blasts were positive (>20% of cells) for CD41 (77.0%) and HLA-DR (43.0%) and were negative for other myeloid and lymphoid markers.
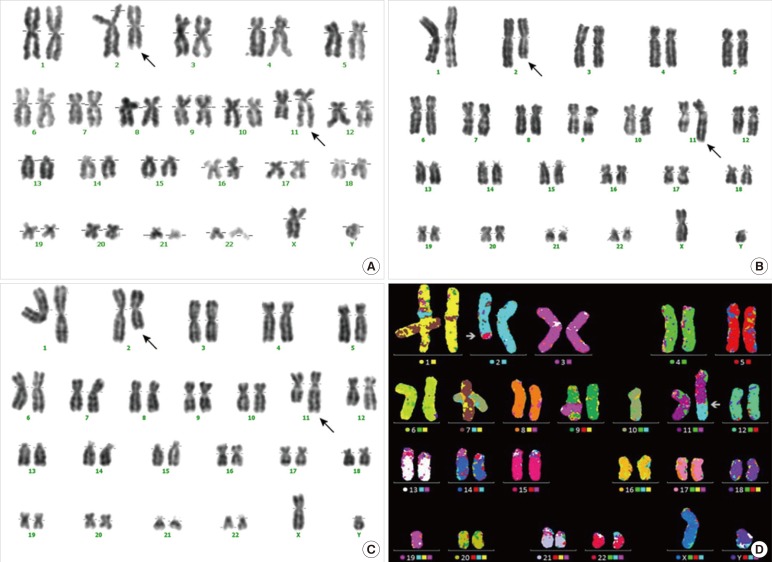
Conventional cytogenetic analysis of the BM aspirate revealed a complex abnormality: 6 out of 20 metaphases were inappropriate, and the karyotype at initial diagnosis of AMKL was 45, XY,-22,t(2;11)(q32;q23),der(14)t(14;22)[6]/46,XY,t(2;11)(q32;q23)[8] (Fig. 1A). He was diagnosed as having AMKL. After receiving induction and consolidation chemotherapy, the patient achieved morphological and clinical remission. At that time, the candidate donor was a sibling 4-yr-old male without a medical history who was matched at high-resolution HLA typing. Donor evaluation was performed according to standard strategies. Subsequently, allogeneic BMT with the matched sibling donor was performed. BM examination on day 124 after the SCT revealed trilineage engraftment and complete chimerism. However, cytogenetic analysis of the BM aspirate still demonstrated the t(2;11)(q32;q23) abnormality (Fig. 1B).
To investigate the possibility of a donor-derived chromosomal abnormality, we performed conventional cytogenetic analysis and M-FISH from peripheral blood (PB) of the donor. Mononuclear cells from the PB of the sibling donor were determined to have a constitutional chromosomal aberration of 46,XY,t(2;11)(q32;q23) [20] (Fig. 1C, D). Follow-up observations were recommended owing to the possibility of hematological malignancies. Fortunately, this patient maintained complete chimerism without evidence of a relapse.
AMKL is recognized as AML-M7 according to the French-American-British (FAB) cooperative group classification system [4, 5, 6]. Childhood AMKL is rare in the general pediatric population. However, it is the most common form of Down syndrome-related leukemia, and its prognosis is favorable in this group of patients [5, 6]. AMKL in the absence of Down syndrome appears to be more heterogeneous, and its prognostic factors are not well defined [3]. The t(1;22)(p13;q13) translocation, forming the chimeric fusion transcript OTT-MAL, is the most common chromosomal abnormality in infants with AMKL not affected by Down syndrome [7]. However, molecular genetic abnormalities in children with AMKL besides trisomy 21 or t(1;22) (p13;q13) are extremely rare.
The transmission of various abnormal karyotypes from phenotypically normal donors after an SCT has been reported previously. Although donor-derived leukemia and myelodysplastic syndrome are rare complications of SCT, several well-documented cases involving constitutional cytogenetic abnormalities of donor cells were reported [8, 9]. Other authors have described cases of relapsed patients showing abnormal post-transplant karyotypes [3, 10].
Thus, it is important to consider the possible risk that a donor chromosome abnormality such as t(2;11)(q32;q23) could be transmitted to the recipient, which could be avoided if chromosomal analysis is conducted during donor evaluation. An additional concern is whether there may be an additional risk to potential donors with conditions such as constitutional chromosomal abnormality when exposed to stem cell stimulants (including granulocyte colony-stimulating factor) during preparation for stem cell harvest.
In summary, we report the case of an AMKL patient with the constitutional chromosomal aberration of t(2;11)(q32;q23). At the time of complete chimerism after SCT, this chromosomal abnormality was persistently found in the BM aspirate of the patient as well as in the PB of the sibling donor. This posed a unique management dilemma for the healthy sibling donor. Owing to the paucity of data, no conclusion could be drawn regarding the possibility of hematologic malignancy in patients carrying the cytogenetic abnormality of the t(2;11)(q32;q23) translocation. Moreover, this case might encourage further discussion about the validity of chromosome analysis for the purpose of BMT or SCT donor evaluations.
 XML Download
XML Download