

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
CHARGE syndrome MIM #214800 is an autosomal dominant genetic disorder with multiple congenital anomalies. The syndrome derives its name from the first letters of its main clinical manifestations: Coloboma, Heart defects, Atresia of choanae, Retardation, Genitourinary malformation, and Ear abnormalities. The clinical criteria for CHARGE syndrome were first described by Blake et al. [1], and then modified by Verloes [2]. According to Verloes' criteria, CHARGE syndrome can be classified as typical, partial, or atypical.
The chromodomain helicase DNA binding protein 7 (CHD7) gene located on chromosome 8q12.1 is 188 kb in length and consists of 38 exons. The CHD7 protein functions as a regulator of DNA transcription [3]. CHD7 gene mutations have been identified in 65-70% of patients with CHARGE syndrome [4, 5, 6]. As most of cases of CHARGE syndrome are caused by a de novo mutation [7], familial cases are rarely reported.
CHD7 gene mutations have been identified throughout the coding exons, and most of them are point mutations. Large deletions and duplications account for only 2% of the observed mutations, while translocations account for <1%. As they are rare, large CHD7 gene deletions and duplications have not been reported previously in the Korean population. Here, we report a typical CHARGE syndrome patient with a large deletion in the CHD7 gene and a presumptive relevant mechanism.
Go to : 
CASE REPORT
A 16-month-old boy was referred for genetic workup because of typical manifestations of the CHARGE syndrome. Facial asymmetry was observed at birth, and auditory and visual dysfunctions were also noted. Heart defects, including aortic stenosis, persistent ductus arteriosus, atrial septal defect, and pulmonary stenosis, were identified on cardiological examination. Multiple abnormal findings, including incomplete cochlear turn and dysplasia of the vestibule and semicircular canal, were found on a computerized tomography scan. Two major signs (coloboma and hypoplastic semicircular canals) and four minor signs (rhombencephalic dysfunction, abnormal middle or external ear, malformation of mediastinal organs, and mental retardation) were identified according to Verloes' criteria. The patient was diagnosed as having typical CHARGE syndrome. A CHD7 gene analysis was requested for a confirmative diagnosis.
Informed consent was obtained from his legal representative, and genomic DNA was extracted from whole blood. PCR was performed by using primers specific for the 37 coding exons of the CHD7 gene. The sequencing reaction was performed with an ABI 3730 analyzer (Applied Biosystems, Foster City, CA, USA) by using a BigDye Terminator v3.1 Cycle sequencing kit (Applied Biosystems). Sequencher 5.0 software (Gene Codes Corporation, Ann Arbor, MI, USA) was used for the sequencing data analysis. No mutations were identified from this CHD7 gene sequence analysis.
Gene dosage analysis was performed by using a multiplex ligation-dependent probe amplification (MLPA) kit (SALSA MLPA P201-C1 CHARGE probemix; MRC-Holland, Amsterdam, Netherlands). MLPA analysis of the CHD7 gene revealed a heterozygous exon 3 deletion (Fig. 1). Genetic analyses of the patient's parents were not available. Further experiments were then performed in order to identify the precise breakpoints of this large deletion. A total of nine Alu sequences were located in intron 2 and three were located in intron 3 (Fig. 2C). Long range PCR was performed by using five forward primers adjacent to AluSx3, AluSg, AluSx1 (the more distal of the two), AluY, and AluSx, and one common reverse primer adjacent to AluSq2. The proximal breakpoint was localized between AluSg and AluSx1 (the more distal of the two). Another long range PCR was performed by using one common forward primer adjacent to AluSg, and two reverse primers adjacent to AluJr and AluSq2. The distal breakpoint was localized between AluJr and AluSq2. An additional long range PCR was performed by using forward primers located 1 kb, 2 kb, and 3 kb distal to AluSg and reverse primers located 1 kb, 2 kb, and 3 kb proximal to AluSq2. The proximal breakpoint was located between 3 kb distal to AluSg and AluSx1 (more distal of the two), while the distal breakpoint was located between 3 kb and 2 kb proximal to AluSq2. Finally, a 1kb-sized PCR fragment was obtained by using the following primers: GGTGGGCTGTGAAGTGTTCTGGC (forward primer; located in intron 2) and ACCCACAGTGCACTCCTCCCC (reverse primer; located in intron 3) (Fig. 2A). Sequence analysis revealed the exact breakpoints (Fig. 2B). The deleted region totaled 38,304 bp from c.1665+10039 in intron 2 to c.2097-3547 in intron 3, and it was accompanied by a TAAC insertion (Fig. 2D).
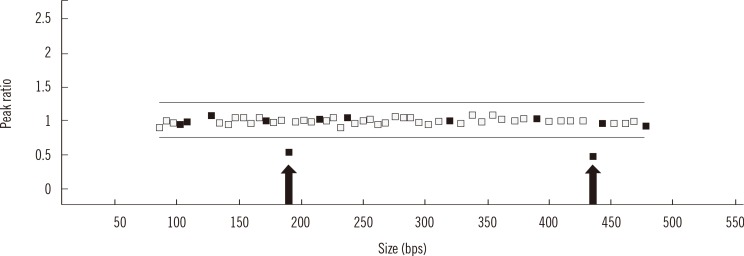 | Fig. 1Multiplex ligation-dependent probe amplification analysis of the CHD7 gene. Arrows indicate the reduced ratio of exon 3.
|
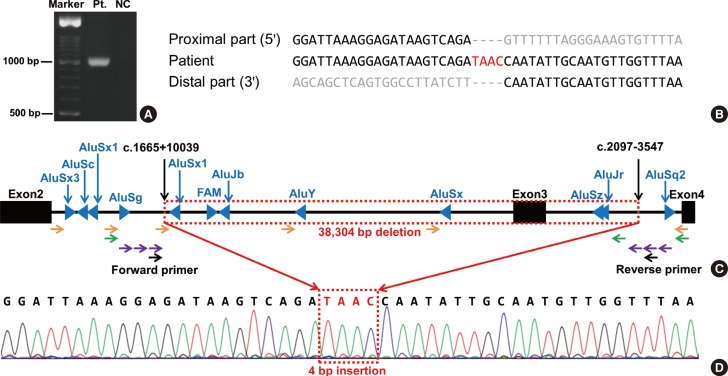 | Fig. 2Results of the sequence analysis and genomic structure encompassing exons 2-4 of the CHD7 gene. (A) Electrophoresis of a PCR product using the forward and reverse primers designated in (C) showed an apparent band in the patient. (B) The proximal and distal parts had no homologous sequences near the breakpoints. (C) Schematic diagram showing the range of the 38,304-bp deletion from c.1665+10039 to c.2097-3547. Alu sequences (blue triangles) were not adjacent to the breakpoints. Orange, green, and violet arrows indicate stepwise long-range PCR primers. (D) Sequence analysis revealed a 4-bp insertion (TAAC) between the breakpoints.
Abbreviations: Pt., patient; NC, normal control.
|
Go to :
DISCUSSION
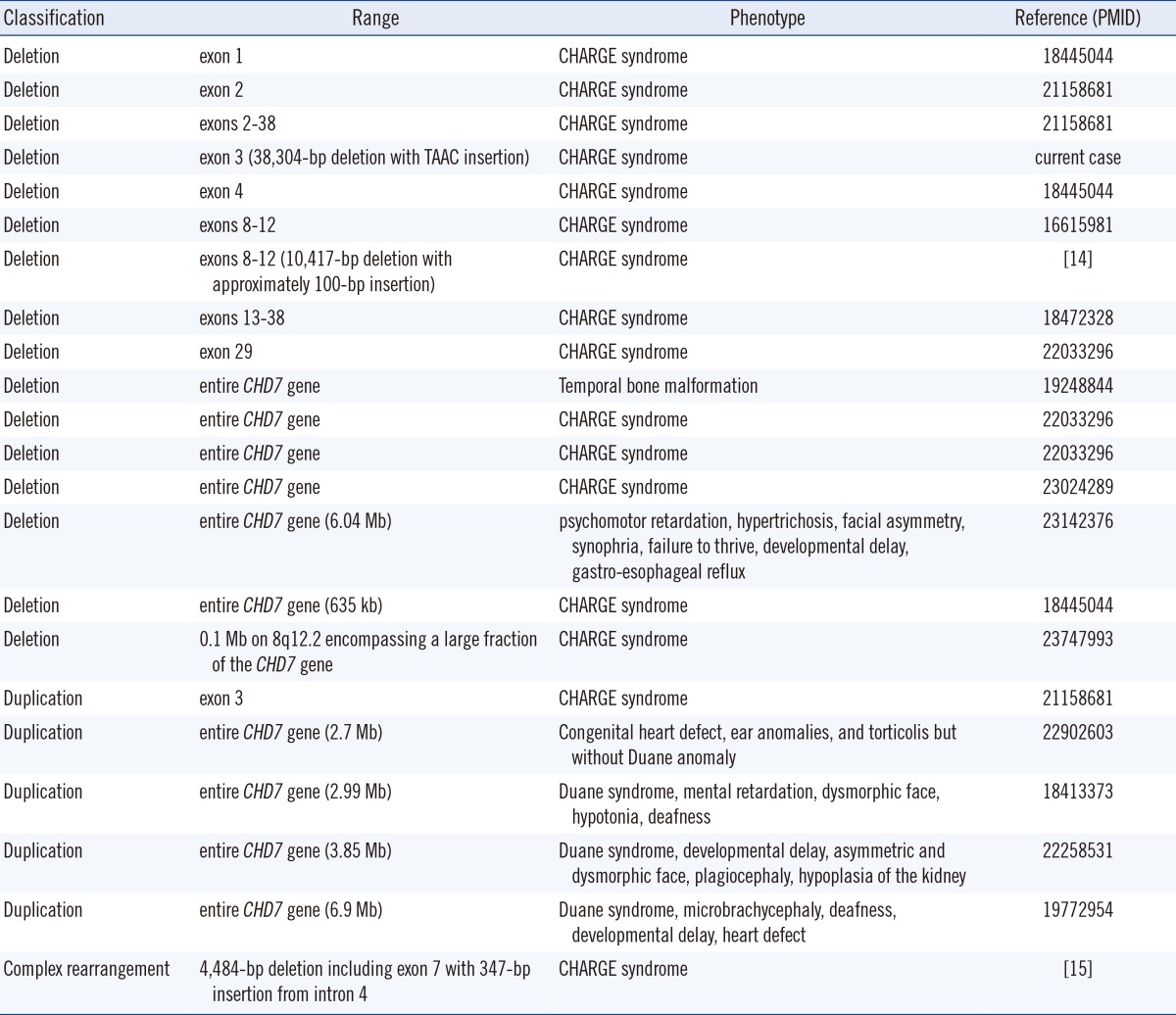
The majority of CHD7 gene mutations are point mutations-nonsense mutations, 44%; frameshift deletions or insertions, 34%; splice site mutations, 11%; and missense mutations, 8% [8]. Large deletions or duplications account for only 2% of total cases. To date, 15 cases of large deletions and 5 cases of large duplications in the CHD7 gene region have been reported (Table 1). Of these 20 cases, 10 cases involved whole gene deletions and duplications. To date, 16 index cases of CHD7 gene mutations have been reported in Korea [9, 10, 11, 12]. However, large deletions and duplications have not yet been reported in the Korean population.
Copy number variations including both additions and deletions are mediated by homologous recombination and non-homologous repair mechanisms [13]. Homologous recombination is composed of non-allelic homologous recombination and single-strand annealing. Non-homologous repair mechanisms are classified into non-replicative non-homologous repair (non-homologous end joining [NHEJ] and microhomology-mediated end joining), and replicative non-homologous repair (replication slippage or template switching, fork stalling and template switching [FosTes], and microhomology-mediated break-induced replication [MMBIR]).
Two different mechanisms underlying CHD7 gene deletions have been previously reported. In the first case, a large deletion spanning exons 8-12 was detected in a Japanese girl [14]. The deletion encompassed 10,417 bases from intron 7 to intron 12. A polyadenine tract of approximately 100 bases was inserted into the junction. Therefore, this deletion was attributed to an Alu retrotransposition-mediated mechanism. In the second case, a complex genomic rearrangement was detected in a Caucasian girl [15]. A deletion of approximately 4,484 bases, including exon 7, was accompanied by an insertion of 347 bases from intron 4 between the breakpoints. This deletion was attributed to a FosTes or MMBIR mechanism.
The CHD7 gene mutations in the current study were mediated by a distinctly different mechanism from the aforementioned cases. Alu sequences were not adjacent to the breakpoints of the deleted region in the current case. The nearest Alu sequences from the proximal and distal breakpoints were located 469 bp and 2,400 bp away, respectively. There was a 4-bp, non-template insertion at the junction site, and there was no sequence microhomology between the upstream and downstream sequences near the junction. Therefore, NHEJ is the most probable mechanism responsible for the current case. NHEJ is a part of the double-strand break repair pathway and is the predominant repair mechanism used in mammals [16]. Furthermore, it is not uncommonly related to large deletions [17]. One or more nucleotide bases can be inserted into the junctions during the repair procedure in NHEJ [18, 19]. In summary, we report the first case of a CHD7 gene deletion mediated by NHEJ worldwide.
Go to :
 XML Download
XML Download