

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mitochondria are maternally inherited and are usually described as the powerhouse of the cell. In addition to their role in the generation of cellular energy in the form of ATP, mitochondria are involved in additional functions, such as signaling, apoptosis, cellular differentiation, growth, as well as cell cycle control. The number of mitochondria in a cell can range from one to several thousand, and this range is determined by the tissue type and organism. Intriguingly, mitochondria possess their own DNA, called mitochondrial DNA (mtDNA), and the extent of alterations that occur to the mtDNA during development affects the presence and emergence of mtDNA mutations in the cell [1, 2].
Human mtDNA is composed of 16,569 nucleotide bases and encodes 13 polypeptides of the electron transport chain, 22 transfer RNAs, and 2 ribosomal RNAs. mtDNA encodes approximately 3% of all mitochondrial proteins and is present in multiple copies-usually 103 to 104 copies per cell. In addition, mtDNA contains a control region termed the D-loop, which contains considerable genetic variations. In fact, the D-loop forms the basis of forensic medicine in human identification and has been a useful tool in molecular anthropological studies on human origins [3, 4].
The origin of mitochondria is unknown; however, one of the most plausible explanations is the endosymbiosis hypothesis. This hypothesis posits that free-living bacteria had colonized proto-eukaryotic cells over the course of evolution, thereby establishing a symbiotic relationship. Primitive eukaryotic cells with intracellular mitochondria capable of metabolizing oxygen possessed selective advantage in an oxygen-rich environment. The electron transport chain within the mitochondria produces far more energy per molecule of glucose consumed, than anaerobic respiration. The oxidative phosphorylation process carried out in the mitochondria produces 38 molecules of ATP, compared to 2 ATPs by anaerobic glycolysis, and facilitates the conversion of toxic oxygen into water, providing those cells with mitochondria a protective biological advantage [5].
However, the disadvantage of oxidative phosphorylation is the formation of reactive oxygen species (ROS), such as singlet oxygen and hydroxyl radicals, which damage cellular components such as lipids, proteins, and DNA. A normally functioning electron transport chain has a 2% ratio of ROS produced for every electron transported. However, under disease or aging conditions, large quantities of ROS are generated, and this can contribute to cellular deterioration, as well as senescence [6].
Compared to the nuclear genome, mtDNA has a modified genetic code and lack introns and protection from histones, resulting in limited repair capacity. The proximity of mtDNA to sites of ROS generation suggests that mtDNA may be more susceptible to mutations compared to nuclear DNA [3]. Accelerated mtDNA mutation rates can result in features indicative of premature aging, consistent with the view that loss of mitochondrial functions is a major causal factor in aging [7]. mtDNA damage and the resultant mitochondria dysfunction has been implicated in a wide range of human diseases, such as constitutional mitochondrial diseases, chronic degenerative/inflammatory diseases, and cancer.
This review summarizes and presents recent publications illustrating the heterogeneity that exists within mtDNA sequences in the Korean population, as well as mtDNA aberrations and their pathological implications in hematological malignancies, solid cancers, chronic inflammatory diseases, and diseases due to environmental hazards such as benzene and polyaromatic hydrocarbons.
HETEROGENEITY OF mtDNA SEQUENCES IN THE KOREAN POPULATION
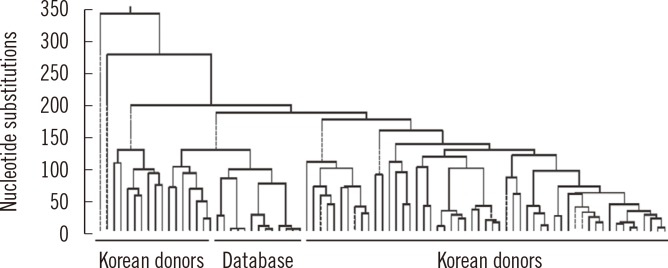
Our laboratory examined sequence variations in the mtDNA control region, transfer RNA leucine1 (tRNA leu1), and cytochrome b (CYTB) genes to investigate the characteristics of mtDNA polymorphisms and haplogroups in the Korean population. Small deletion mutations exist only in the hypervariable (HV) region. A phylogenetic tree revealed a wide range of diversity in mtDNA sequences with 17.7% to 99.9% identification (Fig. 1). The population exhibited a high level of length heteroplasmy (mixture of wild type and mutant sequences) in the 16184-16193 and 303-315 homopolymeric C (poly-C) regions from the mtDNA control region. Some of the most common polymorphisms found in all subjects were 73A>G, 263A>G, 3107delC, and 15326A>G from HV2, HV1, tRNA leu1, and CYTB genes, respectively. The most common haplogroup in the population was D4, found in approximately 16% of the population, followed by A, B, B4a, D5, G1a, and M10 (each 6%). Haplogroup D4 was further categorized into subgroups D4a, D4b, D4e, D4g, D4h, and D4j. However, these data from the Korean population do not correlate with haplogroup distributions described in previous studies and are more similar to the general pattern and frequency of haplogroups of the Japanese population than that of the Han Chinese population. The distinction between polymorphisms, including both common and novel mutations, is also poorly defined. For these reasons, we have strived to build a database for mtDNA sequences representing different age groups in the Korean population [8].
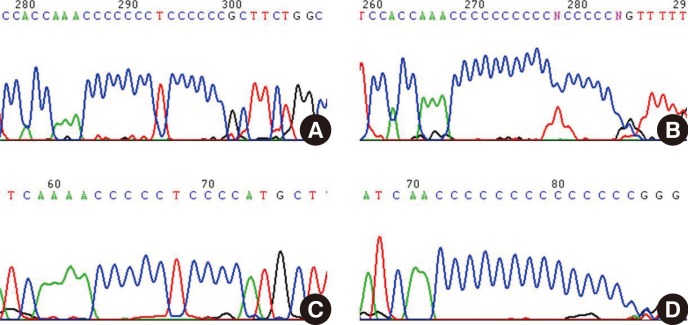
During the analysis of mtDNA HV1 and HV2 sequences from Korean donors, we experienced extreme difficulties when we attempted to sequence beyond the poly-C regions, and therefore we postulated that a possible reason was a high degree of length heteroplasmy (Fig. 2). The length heteroplasmies in the HV regions of mtDNA from blood cells were examined in 70 healthy Korean donors. Interestingly, all subjects displayed length heteroplasmies in both the HV1 and HV2 regions. Closer examination of the HV2 length heteroplasmies indicated that 84% of these donors exhibited a minimal 303-315 poly-C tract frame shift of 1 bp. Sixteen percent of the donors possessed poly-C tract frame shifts of 2 bp or more. The mtDNA copy number in the donor group with major length variants (two or more frame shifts) was about twice as low as in the group with only a 1-bp frame shift. A wide range of mtDNA polymorphisms as well as new sequence variants in each age group were found; however, there was no significant correlation between the number of mtDNA mutations and an increase in donor age. Therefore, these results do not correlate with the hypothesis that the number of mtDNA mutations is in direct proportion to age, or related to the phenomenon of aging. The mutation rate of mtDNA is at least 10-fold higher than that of nuclear DNA. This higher mutation rate is attributed to the lack of protective histones, inefficient DNA repair capacity, proximity of ROS generated by the electron transport chain, and unique structural characteristics that favor mutational events [8].
mtDNA ABERRATIONS IN HEMATOLOGICAL MALIGNANCIES
1. Role of mitochondria in hematopoiesis
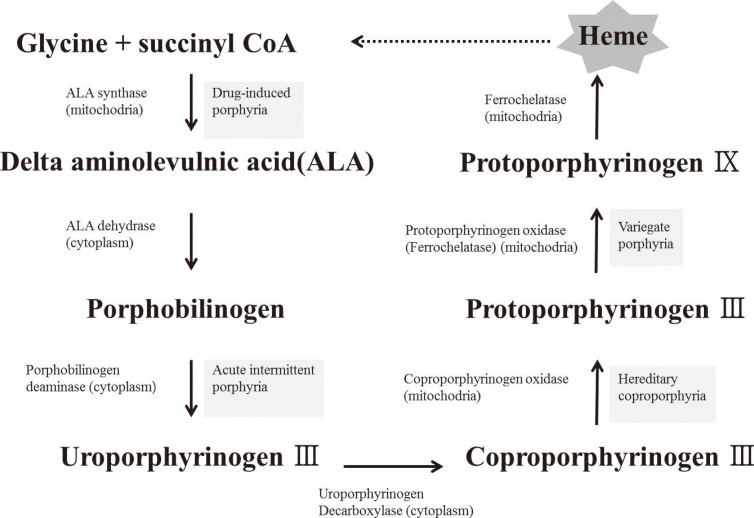
The heme biosynthetic pathway originates in the mitochondria, and after a few intermediate steps in the cytoplasm, is returned to its original location in the mitochondria. It has been speculated that sideroblastic anemia is caused by an enzyme defect in the heme biosynthetic pathway, which leads to a shortage of heme precursors and thereby impairs the utilization of iron that is imported into the mitochondria. In erythroblasts, virtually all the iron that enters the cell via the transferrin receptor enters into the mitochondria, where it is incorporated into protoporphyrin IX to produce heme. Heme is then exported from the mitochondria to combine with globin chains synthesized on cytoplasmic ribosomes [9]. In the case of defective protoporphyrin synthesis, the imported iron lacks its reaction partner and would therefore accumulate in the mitochondria (Fig. 3).
2. Aging of the hematopoietic system and mitochondrial dysfunction
Hematopoietic stem cells (HSCs) have a very high turnover rate; nonetheless, they are not protected from age-related damages. Aging of the hematopoietic system is exhibited through increased incidence of myeloid proliferative diseases, such as MDS and cancer, and through deterioration of the adaptive human immune system. Since HSCs are responsible for sustaining the blood system throughout life, age-related changes could be due to functional deterioration in HSCs. Although HSCs are able to sustain blood production for multiple life cycles, as demonstrated by serial transplantation in mice, phenotypically and functionally they undergo dramatic changes during the aging process [10]. The most profound effect is on the adaptive immune system, where there is a marked decline in lymphoid function in the elderly. Additionally, aging causes overproduction of myeloid cells, which leads to a pro-inflammatory environment [10, 11].
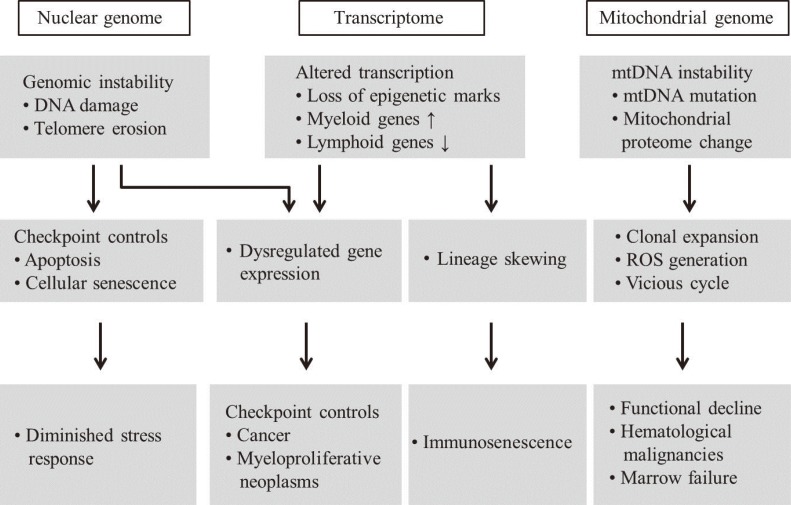
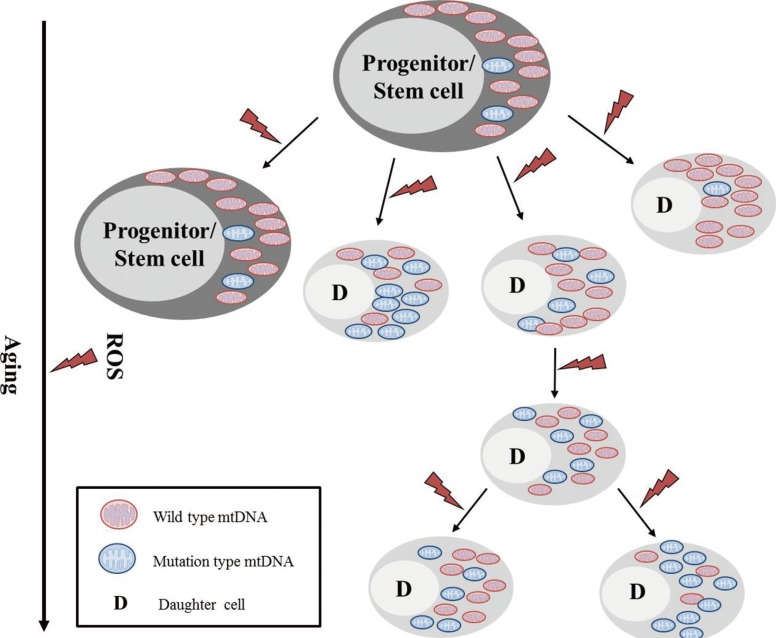
Advanced aging is accompanied by a number of clinically significant conditions arising from the hematopoietic system, including diminution and decreased competence of the adaptive immune system, elevated incidences of certain autoimmune diseases, increased hematological malignancies, and elevated incidences of age-associated anemia. As with most tissues, the aged hematopoietic system also exhibits reduced capacities for regeneration and recovery back to normal homeostasis after injury or stress [10]. The mechanisms underlying aging of the hematopoietic system vary and include intrinsic and extrinsic factors associated with the aging environment that act together to adversely influence the production and functions of hematopoietic effector cells (Fig. 4). However, increasing evidence suggests that age-dependent cellular and molecular alterations within the most primitive hematopoietic stem cell compartments may significantly contribute to hematopoietic deterioration during aging [12].
The classical pathway for the mitochondrial theory of aging is that by-products of mitochondrial phosphorylation (ROS: superoxide anion, hydrogen peroxide, and hydroxyl radicals) results in damages to mitochondrial macromolecules including the mtDNA. When these factors impair the apparatus for mitochondrial energy generation beyond a functional threshold, proteins are released from the mitochondria that activate the caspase pathway and lead to apoptosis. The aging process can introduce excessive mutations in the mtDNA via error-prone polymerase γ, which in turn can sufficiently impair mitochondrial functions, without causing further increase of ROS [13, 14, 15].
It has been suggested that while somatic mtDNA mutations accumulate with age in post-mitotic tissues, the mutations are diluted and lost in continuously proliferating tissues such as the bone marrow. However, on average, 25% of individual CD34+ clones from the adult bone marrow showed mtDNA heterogeneity or sequence differences from the aggregate mtDNA sequences of total bone marrow cells from the same individual. In contrast, only 1.6% of single CD34+ clones from cord blood showed mtDNA sequence variation from the aggregate pattern. Thus, it appears that the age-dependent accumulation of mtDNA mutations appears relatively frequently in mitotically active human tissues, and thus poses important implications in the aging process in hematopoietic tissues (Fig. 5) [13, 14].
3. Mitochondrial aberrations in bone marrow failure syndromes
Bone marrow failure syndromes encompass a group of disorders that are either inherited or acquired. These diseases are HSC disorders that can involve all cell lines: erythroid for red blood cells, myeloid for white blood cells, or megakaryocytic for platelets. The MDS are a heterogeneous group of hematological diseases characterized by bone marrow failure and are associated with increased risk of malignant transformation. Cytogenetic abnormalities are highly prevalent and in fact correlate with the prognosis and progression of leukemia [2]. The more aggressive categories of MDS, especially from secondary exposure to alkylating drugs, topoisomerase inhibitors, and radiation, share similar risk factors with acute leukemia and show stereotypical chromosomal abnormalities.
Functionally relevant point mutations were found in the mitochondrial RNA and polypeptide-encoding genes of 50% of the patients with MDS. Their increasing mutation load correlates with MDS and the development of AML. Several point mutations for Leber's hereditary optic neuropathy occur in the bone marrow and may exert a synergistic effect on bone marrow stem cells via the apoptotic pathway. We systematically analyzed the entire mitochondrial genome by gene amplification and direct sequencing of samples from 10 patients with MDS. Overall, no increase was observed in the number of mtDNA genes harboring polymorphisms or mutations in the patients examined, compared to 8 healthy controls; however, there were a few more mtDNA changes as a result of amino acid alternations in MDS. Thirty novel mutations-all nucleotide substitutions-were found distributed throughout the mitochondrial genome among the 10 patients, with 5 mutations resulting in amino acid alternations. None of the mutations in the control group produced amino acid alterations. We were unable to confirm previously described mutations in sideroblastic anemia or 'hot spots' in the cytochrome c oxidase (CO) I and II genes. Our data indicated that mitochondrial genomic instability does not play a major role in MDS and also does not correlate with previous reports of significant or widespread mitochondrial mutations in this disease [2]. Modest changes in mutation numbers and mitochondrial microsatellites may be evidence of increased mutagenesis in mtDNA, or a reflection of limited clonality among HSCs in the bone marrow failure syndrome [2, 16].
Aplastic anemia is considered a heterogeneous disease based on the numerous different putative etiologies, such as idiopathic, secondary causes (radiation, drug and chemicals, viruses and immune diseases), paroxysmal nocturnal hemoglobinuria, and pregnancy [16]. Complete mtDNA nucleotide sequences were analyzed in bone marrow specimens taken from 9 Korean patients who were afflicted with aplastic anemia, as well as 8 healthy individuals. Several polymorphisms and novel mutations were found throughout the entire mtDNA genome in both the patient group and the control group. In patients with aplastic anemia, 12 mutations produced amino acid alterations; however, none of the mutations found in the control group produced amino acid alterations. More heteroplasmic mutations and nonsynonymous mtDNA changes were observed in patients with aplastic anemia. The number of mtDNA aberrations in bone marrow cells from patients with aplastic anemia (25.6±14.3, mean±SD) was significantly higher compared to that in controls (12.8±7.3) (P=0.019). These data suggested an association between mtDNA aberrations and aplastic anemia [16].
4. mtDNA aberrations in leukemia and leukemia stem cells
mtDNA can be easily damaged owing to the lack of histone proteins, inefficient repair ability, and close vicinity to the respiratory chain and ROS production [17]. mtDNA damage is more extensive and persists longer than nuclear DNA damage [18]. Pathogenic mutations can affect certain mtDNA copies in a cell, resulting in heteroplasmy (combination of mutated and wild type mtDNA) or homoplasmy (uniformity of sequences within an individual cell), which affects the entire mtDNA repertoire within the cell. The somatic mutation process in leukemia is complex, leading to diverse levels of genetic alterations due to either an intrinsic aspect of leukemia pathophysiology or chemotherapy effects [19]. Thus, it appears that somatic mutations may accumulate at varying rates in differentiated cells and progenitor cells from the same individual [19]. At diagnosis, some leukemic patients present mtDNA alterations that were comparable to controls. The mtDNA sequence is relatively homogenous in CD34+ cells from cord blood, and much more heterogeneous in CD34 cells from adults aged 25-57 yr. The observed complex pattern in leukemia patients suggests that mutations emerge at a relatively high rate in leukemic cell mtDNA, with homoplasmy and clonal expansion (expression of the fixed mtDNA mutation in a substantial proportion of progeny) occurring over months rather than decades [19].
5. mtDNA aberrations in acute myeloid leukemia
Numerous novel mtDNA mutations were found only in primary AML cells (unpublished data). This observation provided insights into the analysis of mitochondrial proteins and development of novel chemotherapeutic agents. Aberrant mitochondrial protein expression was found in primary AML cells, and this could prove to be helpful in the identification of new molecular biomarkers and the development of novel anti-cancer agents. The mitochondrial protein alteration in primary AML cells imply that the mutation process in mtDNA is increased due to the high level of oxidative stress that occurs during ATP synthesis
Recent studies suggested that mitochondrial genomic instability (mtGI), which consists of substitutions, deletions, length heteroplasmic mutations, and mtDNA copy number alteration, is observed in primary AML cells. As a result of mtGI, enzyme activities of the mitochondrial respiratory chain complexes are reduced. Our data indicated that many mutations and polymorphisms (a total of 606 mtDNA sequence variants) identified were from the bone marrows, buccal mucosa, and blood samples of 48 patients with AML.
6. mtDNA aberrations in acute lymphoid leukemia
By comparing entire sequences of the mitochondrial genomes of normal buccal epithelium and leukemic cells, a recent study found that 4 out of 6 patients (67%) with adult-onset ALL harbored leukemic cell-specific mtDNA mutations [20]. Bone marrow samples taken from ALL patients at different stages of the disease indicated that the A15296G mutation, as observed in the presentation and relapse of bone marrow samples, is a clonal marker for ALL, making it a suitable biomarker for the detection and monitoring of the disease during treatment. Not many studies have been conducted on mtDNA aberrations in ALL; therefore, additional investigation is necessary to reveal the spectra of mtDNA mutations and their corresponding pathophysiological and clinical implications.
Polymorphisms around the H-strand replication origin (nucleotides 150 to 199) and conserved sequence block II (nucleotides 299 to 317) in mitochondrial genes were associated with leukemia biology and treatment response. T-cell ALL patients were more likely to exhibit high levels of length heteroplasmies at the nucleotide position 303 poly-C tract. The T199C polymorphism was associated with increased risk for ALL in the Malaysian population [20]. Patients with the T152C polymorphism showed better treatment responses than those patients without the polymorphism. No differences were observed in mtDNA contents between diagnostic ALL samples and controls; however, there was a significant reduction in mtDNA content after treatment, especially in patients with polymorphisms located in the origin of H-strand replication. Somatic mutations were found in 13% (9 of 76) of patients, suggesting an association to leukemogenesis. This study suggested that polymorphisms affecting transcriptional control affect mtDNA replication. In childhood ALL, decreased mtDNA copy number after treatment may confer susceptibility to chemotherapy [20].
7. mtDNA aberrations in chronic lymphocytic leukemia
In both untreated and previously treated CLL patients, several nucleotide alterations were identified in the D-loop region. The role of mtDNA mutations in oncogenesis and chemosensitivity is unclear; however, increases in mtDNA mutations were observed during ROS generation. mtDNA mutations in primary leukemia cells are caused by DNA-damaging chemotherapeutic agents, resulting in heteroplasmy. After chemotherapy, new heteroplasmic mutations (G-C/G), identified by sequencing analysis, altered the CO II gene at nucleotide position 7762 [21].
Mitochondrial defects have been implicated in the development and progression of cancer for several decades. The groundbreaking work by Warburg illustrated that cancer formation is precipitated by damage to the respiratory machinery, which results in an increase in glycolytic ATP production as compensation; some researchers claim that the observed effects are associated with mtDNA mutations [21].
The mitochondrial respiratory chain produces intracellular superoxide radicals, and ROS generation is known for causing mtDNA mutations. Due to this biological consequence, patients who had undergone chemotherapy exhibited increased superoxide generation in CLL cells and increased mtDNA mutations. In conclusion, instabilities among B-CLL patients demonstrate neutrality to DNA functions and likely do not promote tumorigenesis [22].
8. Mitochondrial aberrations in leukemia stem cells
Cancer stem cells have recently been discovered in colon cancer and brain tumors, and they have been shown to demonstrate resistance against anti-cancer drugs and radiotherapy. Thus, it is imperative that novel drugs and treatment strategies that can effectively target cancer stem cells are identified and developed. Leukemic stem cells (LSCs) may play a pivotal role in the pathogenesis of hematological malignancies such as AML. Due to the slow division process and long interphase of LSCs as compared to normal stem cells and hematopoietic cells, many anti-cancer therapeutics are not effective in LSC treatment. Our laboratory has been examining the growth and proliferation capacity (plating efficiency) of clonogenic hematopoietic progenitors and LSCs, by comparing healthy donors and AML patients by using single cell sorting and various culture systems (BD FACS Aria cell sorter; BD Biosciences, San Jose, CA, USA).
A total of 384 normal hematopoietic stem cells (CD34+ CD38+/CD38-) were obtained from the peripheral blood and cord blood donated by four donors, using a single cell sorter. Individual single cells were cultured in 96-well plates with each well containing 100 µL of serum media, 100 ng/mL of stem cell factor, 100 ng/mL of Flt-3, 100 ng/mL of thrombopoietin, and 50 ng/mL of G-CSF for five days. There were 768 single CD34+ CD38- LSCs and 384 single CD34+CD38+ cells obtained from 3 AML patients. The growth and proliferation capacities of normal HSCs and LSCs were determined in terms of plating efficiency (number of wells in which more than two cells had proliferated/ total number of cells in 96-well plate culture×100). The plating efficiency demonstrated by an individual's normal single HSC varied between samples. Approximately 88 out of 192 single stem cells from healthy donors originated from cord blood, and propagated to more than 2 cells with a plating efficiency yield of 45.8% and 30.2% (58/192). In contrast, a single LSC from AML patients showed significantly lower plating efficiency with 14.6% (42/288), 3.6% (7/192), and 8.0% (23/288). These results directly corroborated the properties of quiescence and slow division exhibited by LSCs. In addition, the plating efficiency of normal HSCs was shown to vary between healthy donors (unpublished data).
Single AML stem cells exhibited significantly lower plating efficiency, implying the properties of quiescence and slow division exhibited by the cells. AML stem cells possessed significant alterations of length heteroplasmy in the mtDNA control region, which may lead to a decline in mitochondrial biogenesis (reduction of mtDNA copy number) and dysfunction of mitochondrial ATP synthesis. These findings supported the introduction of aggressive compensatory mechanisms by the mitochondria of AML stem cells, and also implied potential strategies for the development of novel LSC-targeting therapeutics.
9. mtDNA aberrations in non-Hodgkin's lymphoma
The mtDNA copy number was increased in patients with CLL, Burkitt lymphoma, Epstein-Barr virus-transformed lymphoblastoid cell lines, and T-cells activated via the T-cell receptor. It was recently reported that the fact that peripheral white blood cells possessed higher mtDNA copy numbers as compared to healthy individuals is associated with future risk of non-Hodgkin's lymphoma (NHL). There exists a dose-response relationship between tertiles of mtDNA copy number and the risk of acquiring NHL (odds ratio [OR], 95% confidence interval [CI]: 1.0; 1.4 [0.7-2.8]; and 2.4 [1.0-5.5], respectively; P (trend)=0.046). The effect was most pronounced for the CLL/small lymphocytic lymphoma (SLL) subtype (OR: 1.0; 3.2 [0.7-15.7]; 14.1 [1.9-103.2]; P (trend)=0.009). These results suggested a correlation between mtDNA copy number and the risk of NHL, particularly CLL/SLL [23].
10. mtDNA sequence alterations and new therapeutic targets in multiple myeloma
Cancer cells are more prone to harbor altered mitochondrial electron transport chains compared to normal cells, leading to a state of metabolic oxidative stress and in turn more efficient for ROS production [24].Therefore, it follows that therapies which can specifically target elevated ROS production in cancer cells should be able to induce apoptosis. Examples of such agents are anti-cancer drugs which target mitochondria [24, 25].
To study mtDNA aberrations in multiple myeloma (MM) cells, we investigated the rates of mtDNA mutations in MM cells from bone marrow aspirate specimens at the initial diagnosis. mtDNA mutations were detected exclusively in the CD133+ myeloma cells, but not in the CD33+ bone marrow cells from the same patients.
Using total bone marrow cells from 6 MM patients,CD133+ MM cells and CD33+ cells were used as normal internal controls and the isolation of both was performed using a single cell sorting system (FACS Aria cell sorter, BD Biosciences). Highly selective cell sorting procedures were used to target cell populations with unique expressions of CD133 and CD33 markers. A total of 3 out of 6 patients (50%) displayed MM cell-specific mtDNA mutations in the HV segment of the control region as length heteroplasmic and substitution mutations, which were not found in the corresponding CD33+ cells from the same patients. MM cell-specific length heteroplasmic mutations were detected in np 16184-16193 poly-C track (two patients) in the HV1 region and np 12385-12391 poly-C in the ND1 gene. These findings suggest the potential of developing new molecular markers for minimal residual disease monitoring, and could be used as new therapeutic agents to target mitochondria in MM and provide new insights into MM mitochondria-related molecular pathophysiology.
11. Usefulness of mtDNA minisatellite markers after allogeneic stem cell transplantation
Chimerism analysis is an alternative technique for monitoring minimal residual disease. However, timely therapeutic intervention is critical to prevent possible relapse or rejection. It is therefore imperative to closely monitor chimerism after allogeneic stem cell transplantation (SCT). Short tandem repeat (STR) markers are commonly used because of the high polymorphic nature between individuals. The STR assay generates quantitative results by using fluorescence-labeled primers and a capillary electrophoresis system. However, when the target DNA content is low or degraded, STR assays are imprecise and have low sensitivity. Careful approach and consideration is required because of this potential increase in imprecision, particularly with a small proportion of donor cells. To circumvent these problems, 6 mtDNA minisatellite (mtMS) markers were developed based on our publications describing the mtDNA genetic diversity among the Korean population. These markers were constructed to monitor the extent of donor cell engraftment in SCT recipients, and also to evaluate their usefulness and sensitivity compared with the nuclear DNA markers [26].
The selected mtMS markers were assessed by using clinical samples and in vitro mixing to test the usefulness, sensitivity, and precision. Chimerism studies were carried out between 14 and 114 days after SCT by using 7nuclear DNA markers (STR-D12S391, VNTR-D1S80, STR-D18S51, STR-F13A1, STR-HUM FABP2, STR-HUM RENA-4, and STR-Amelogenin) and 7mtMS markers, including direct sequencing of the mtDNA HV region. The 303 poly-C and 16184 poly-C mtMS proved to be useful as nuclear DNA D12S391 and D1S80 markers. Meanwhile, the mtMS markers showed a higher sensitivity than the nuclear DNA-STR markers, possibly due to preferential amplification of mtDNA. After allogeneic hematopoietic stem cell transplantation (allo-HSCT), mtMS markers were used in multiple capacities for monitoring mixed chimerism and prognosis. By analyzing the 303 poly-C mtDNA marker and nuclear D18S51 STR marker using variable mixed blood cell concentrations, mtMS markers presented higher accuracies' sat monitoring mixed chimerism than nuclear STR markers, especially in unrelated transplantations and under inappropriate sampling conditions. Longitudinal follow-up after allo-HSCT revealed that chimerism precisely reflected the status of engraftment or relapse during the clinicopathological course. Moreover, changes in mtMS marker levels in recipients before allo-HSCT were associated with detrimental clinical outcomes. The mtDNA copy number per cell is relatively high, and sequencing the mtDNA is easier than the diploid nuclear DNA because of the haploid nature of mtDNA, which leads to preferential amplification.
Moreover, mtDNA typing is used primarily in cases where the nuclear DNA is too degraded or cannot be recovered in sufficient quantities for sequencing to proceed. These facts provide plausible reasons as to why the mtMS markers showed higher sensitivities and are considered more useful than the nuclear DNA markers [27].
mtDNA ABERRATIONS IN SOLID CANCERS
1. mtDNA aberrations in colorectal cancer
Most studies on mtDNA mutations in colorectal cancer lack clinical relevance and include only case-control and case-database comparisons. Our previous work investigated tissue-specific mtDNA mutations in colorectal cancer, and evaluated their clinical relevance from a collection of 54 matched colorectal cancer and adjacent normal tissue samples [28]. Over a half of the patients (59%) harbored cancer tissue-specific mtDNA mutations. The patterns of mtDNA mutations encompass substitutions (13%), mtDNA minisatellite instability (mtMSI) (20%), and both mutations combined (26%). mtMSI in colorectal cancer occurred mainly in the 303 poly-C (35%) and 16184 poly-C (19%) minisatellites. mtDNA copy number and hydrogen peroxide level were significantly increased in colorectal cancer tissues [28]. The numbers of mtDNA large deletions significantly declined in colorectal cancer tissues compared with those from matched normal mucosa (P=0.03). The activities of the mitochondrial respiratory chain enzyme complexes I, II, and III in colorectal cancer tissues were reduced. In addition, the mtDNA haplogroup B4 may be closely associated with colorectal cancer risk. The patient group harboring cancer tissue-specific mtDNA mutations showed larger tumor sizes (P=0.005) and more advanced TNM stages (P=0.002). Thus, mtDNA mutations in colorectal cancer may evoke risk factors that induce negative outcomes and in turn promote tumorigenesis [28].
The numbers of mtDNA large deletions decreased significantly in colorectal cancer tissues compared with those from matched normal mucosa. These findings implied an active selection against mtDNA with the 4,977 bp deletion, or represent a population of tumor cells that does not undergo the typical aging response and subsequent tumor formation [28].
mtDNA haplogroups are frequently found in various cancers, metabolic diseases, aging, and some neurodegenerative diseases. Patients with the mtDNA haplogroup M exhibited increased risks for developing breast cancer. Additionally, the mtDNA haplogroup D4a was associated with increased risks of thyroid cancer [28]. On the other hand, no significant correlations between any mtDNA haplogroups and colorectal cancer were found. However, this study remarked that the mtDNA haplogroup B4 may be closely associated with risks of colorectal cancer among the Korean population. Nevertheless, the number of patients included in our study was limited, and thus further investigation which includes a larger cohort is warranted to elucidate the correlations between mtDNA haplogroups and colorectal cancer risk.
Furthermore, our previous study investigated clinicopathological values according to the existence of mutations, as well as specific mutation types [28]. Patients who harbor both the substitution and mtMSI showed larger tumor sizes and advanced TNM stages, compared to patients with only one type of mutation (substitution or mtMSI) or no mutations at all. In conclusion, owing to the elevated levels of ROS in cancer tissues, most colorectal cancer patients harbored cancer tissue-specific mtDNA mutations, specifically, a type of substitution and minisatellite alteration. mtDNA mutations may be associated with advanced stages, and is a potential risk factor indicative of poor treatment outcome and prognosis in colorectal cancer [28].
2. mtDNA aberrations in bladder cancer
The average mtDNA copy numbers in urine samples and the corresponding peripheral blood samples (83.45±60.36 and 39.0±24.38, respectively) (mean±SD) differed significantly (P<0.001) from our previous study on bladder cancer [29]. The mtDNA copy numbers in the urine samples from patients with high-grade and low-grade bladder tumors (81.83±67.78 and 86.49±46.69, respectively) did not differ significantly (P=0.589). The mtDNA copy numbers in urine samples were much higher than those in the corresponding peripheral blood samples. mtDNA mutations were present in 80% of the D-loop regions in bladder cancer patients. This report further supported the significance of genetic alterations in urothelial bladder carcinoma, and the clinical utility of mtDNA quantitative polymerase chain reaction, mtDNA sequencing, and capillary electrophoresis for diagnostic purposes in patients with bladder cancer [29].
mtDNA ABERRATIONS IN CHRONIC INFLAMMATORY DISEASES
1. mtDNA aberrations in Barrett's esophagus
Carcinogenesis is a long-term, multistep process driven by genetic and epigenetic changes in susceptible cells, which gain a selective growth advantage and undergo clonal expression. Barrett's esophagus (BE) is one of the most common premalignant lesions and can progress to esophageal adenocarcinoma (EA). With respect to morphology, the carcinogenetic process of Barrett's mucosa progresses through increasing grades of epithelial dysplasia. At the present time, intraepithelial neoplasia, also called dysplasia, is the only marker that can be used to define patient populations who are at high risks of cancer. During the malignant transformation from BE to dysplasia and EA, prevalence of these molecular changes is increased. Although for the majority of identified genes, the predictive or prognostic value remains unclear. Nearly all of the new markers have not yet been validated in prospective controlled or randomized studies [30].
Even though many molecular events take part in the neoplastic transformation of Barrett's mucosa, only a few (i.e., changes in DNA ploidy, increased proliferation, and alteration of the p53 gene) have been identified as potential promoters of carcinogenesis. However, the molecular mechanism underlying the progression from BE to EA remains to be elucidated, while most studies on mtDNA mutations in BE have been only performed with dysplasia, and there the data is insufficient. We investigated new molecular events (BE tissue-specific mtDNA alterations/instabilities) in the mitochondrial genome and causative factors for their alterations using the corresponding adjacent normal mucosal tissues (NT) and Barrett's esophageal tissues (BT) from 34 patients afflicted with Barrett's metaplasia but present no dysplasia. Eighteen patients (53%) exhibited mtDNA mutations that were not found in the adjacent NT. The mtDNA copy number was approximately 3 times higher in BT than in the adjacent NT. In fact, the activities of the mitochondrial respiratory chain enzyme complexes were impaired in the Barrett's metaplasia tissues that did not present dysplasia. ROS levels in BT were significantly higher than the corresponding NT samples. Therefore, it is suggested that high levels of ROS in BT can cause mtDNA mutations, which play an important role in disease progression and tumorigenesis in BE [30]. Oxidative damage has long been correlated to mucosal damage of the gastrointestinal tracts and their ensuing carcinogenesis. Furthermore, despite treatment with anti-secretory medications for reflux esophagitis, many patients failed to achieve complete mucosal healing, and instead suffered from sustained symptoms or development of BE. This implied that additional damaging factors or impaired mucosal resistance is involved in the pathogenesis.
Chronic inflammation that is mainly induced by the gastroesophageal reflux disease is the major cause of BE. An increase in the level of ROS production frequently occurs in chronic inflammatory cells and tissues. Thus, the current study detected BT-specific mtDNA alterations under the assumption that mtDNA aberrations are caused by abundant production of ROS in chronically inflamed BT. mtDNA mutations were detected exclusively in BT samples but not in the adjacent NT samples. These mtDNA mutations were observed as base substitutions and length heteroplasmies in the control regions. Oxidative stress elicited by chronic inflammation increases the number of mtDNA mutations in BT, and may correlate with a precancerous status. The levels of ROS were significantly higher in the BT supernatants compared to those from the adjacent NT. This high level of ROS damages the mitochondria, leading to mtDNA mutations. mtDNA control regions known as 'hot spots' of gene mutations contain the mtDNA production-regulating fraction and the HV regions, because mutations at these locations can cause various degenerative diseases and tumors [30].
Importantly, mutations in the control regions may alter the rates of DNA replication by modifying the binding affinities of significant transactivating factors. These mtDNA alterations in BT may further impair the respiratory chain defect and result in an increase in mtDNA copy number to compensate for the ATP deficiency. During this perturbation, mitochondria may produce a large quantity of ROS, which causes the vicious cycle observed in other chronic inflammatory diseases [31]. BT-specific mtDNA mutations frequently occur in both the mtDNA control and minisatellite regions due to excessive production of ROS. High levels of ROS in BT may contribute to the development of mtDNA mutations, which would play a crucial role in the pathophysiology of BE and further progression to EA. Although many risk factors are considered to be attributable to BE and EA, antioxidant treatments appear to be the preferring therapeutic option that is primarily used for prevention or treatment of BE, and can act in a preventive capacity in disease progression [30].
2. mtDNA aberrations in nasal polyps
We investigated, in 23 patients, the possibility that mtDNA mutations promoted inflammatory or chronically damaged nasal polyp tissues. There were 13 patients (57%) who displayed nasal polyp tissue-specific mtDNA mutations in the HV segments of the control regions as well as the CYTB gene, which were not found in the corresponding blood cells and adjacent normal tissues. In np 303-315 homopolymeric poly-C track (39%), np 514-523 CA repeats (17%), and np 16184-16193 poly-C track (30%) nasal polyp tissue-specific length heteroplasmic mutations were also detected. The average mtDNA copy number was approximately 3 times higher in nasal polyp tissues compared to the corresponding peripheral blood cells and adjacent non-polyp tissues. The level of ROS was significantly higher in the nasal polyp tissues compared to the corresponding control samples. Increases in the level of ROS in nasal polyp tissues may be attributable to the mtDNA mutations and contribute to the vicious cycle of the pathophysiology of nasal polyps [31]. A nasal polyp is a relatively common affliction, although the underlying mechanism is currently unknown. A number of causes have been suggested, including chronic inflammation, resistance to aspirin, pollution, damaged epithelium, damaged DNA, and food allergies. Various studies have reported dissimilarities in gene expression in unaffected tissue and nasal polyp tissues [31].
ROS is commonly released in chronic inflammatory tissues. These ROS and free radicals target the mtDNA. With inflammation and excess stress, ROS production can overwhelm the antioxidant system, resulting in mtDNA damages. Nasal polyps most often accompany chronic inflammation, such as the accumulation of focal exudates in tissues, and the proliferation of submucosal tissues in nasal and paranasal sinuses. In conclusion, heteroplasmic mtDNA mutations in nasal polyp tissues frequently occur in both the mtDNA control and coding regions as base substitutions in coding and non-coding regions, and as base insertions/deletions within the control regions in the poly-C tract. The high levels of ROS produced by nasal polyp tissues may induce mtDNA impairment, which can lead to mtDNA mutations. Importantly, mutations in the control regions may alter the rate of DNA replication by modifying the binding affinity of significant transactivating factors. These mtDNA alterations in nasal polyp tissues may further damage the respiratory chain and increase mtDNA copy numbers in order to compensate for the ATP deficiency. During this perturbation, mitochondria may produce a large quantity of ROS, which can cause the vicious cycle observed in other chronic inflammatory diseases [31].
3. mtDNA aberrations in Helicobacter pylori-peptic ulcer
As a result of Helicobacter pylori infections, ROS are commonly released in the inflamed gastric mucosa. It is postulated that mtDNA mutations arise in inflamed or chronically damaged gastroduodenal epithelial cells. Results had shown that mtDNA mutations associated with H. pylori infections occurred in both the mtDNA control and coding regions in peptic ulcer tissues. Approximately half of the patients who were examined presented heteroplasmic mtDNA mutations. The high levels of ROS generated by H. pylori infections caused mtDNA damages leading to mtDNA mutations in peptic ulcer tissues. To compensate for the deficiency in ATPs, the mtDNA copy number is increased. During this perturbation, mitochondria produce a large quantity of ROS, which causes the vicious cycle observed in peptic ulcer diseases [32].
4. mtDNA aberrations in chronic atrial fibrillation
Many studies had reported that various types of somatic mtDNA mutation were found in cardiac tissues. We showed that mtDNA mutations, including small deletions and tissue-specific length heteroplasmic mutations, occurred in both the control and coding regions of mtDNA from patients with chronic atrial fibrillation (cAF). Oxidative injury and deletion of mtDNA in the cardiac muscles are elevated in patients with cAF, which may contribute to the impairment of the bioenergetic function of mitochondria, and the induction of the vicious oxidative cycle attributable to the pathogenesis of atrial myopathy in cAF patients. In conclusion, 9 novel mutations were found in the control and coding regions of mtDNA. Interestingly, 2 patients (50%) had tissue specific to length heteroplasmic mutations from 16184 poly-C and CA repeats, starting at nucleotide 514, which were not found in blood cells. A 9-bp deletion around the mtDNA CO II gene was also identified in the tissues and blood cells from the patients examined. These findings strongly suggested that somatic mtDNA mutations are associated with cAF. Rapid atrial depolarization in cAF may result in higher oxygen consumption and oxidative workload, thus accelerating aging by accumulating somatic mtDNA mutations [33].
MITOCHONDRIA AND ENVIRONMENTAL MEDICINE
Benzene damages the bone marrow and causes a reduction in red blood cells, leading to anemia. It can also cause excessive bleeding and compromise the immune system, thus increasing chances of infection. Benzene causes leukemia and is associated with other blood cancers and pre-cancers of the blood [34]. Benzene directly causes an increase in the generation of intracellular ROS, which subsequently induces changes in mitochondrial mass, mtDNA content, and the proteome. In addition, benzene also causes oxidative stress, leading to an increase in mitochondrial mass and mitochondrial membrane potential.
Benzene exposure in humans and animals has been shown to result in structural and the amount of chromosomal aberrations that occur in lymphocytes and bone marrow cells, indicating that benzene is a genotoxin. The mitochondrial genome is highly vulnerable to DNA damage caused by ROS and mutagens, and exhibits higher mutation rates in comparison to the nuclear genome. Furthermore, the lengths of time it requires to correct DNA damages are much longer in the mitochondrial genome. A number of factors contribute to the vulnerability of mtDNA, including the absence of histones, which provide packaging and protection of nuclear DNA, and the error-prone replication and repair systems of mitochondrial genes. Therefore, our previous work had targeted the mitochondrial genome and proteome to identify biomarkers associated with benzene exposure [34]. Results of the present study showed that after short-term benzene exposure, the mitochondrial mass and mtDNA copy number increased. These findings are consistent with a previous report which showed an increase in mtDNA content in the tissues of aged rats and elderly human individuals. It was considered that during oxidative stress, the number of mtDNA increased in order to compensate for declining respiratory functions; however, the molecular mechanism underlying the increase in mtDNA associated with decreased respiratory functions under aging and oxidative stress conditions remains unclear.
The mitochondrial membrane potential reflects the pumping of hydrogen ions across the inner membrane during the process of electron transport and oxidative phosphorylation-the driving force behind ATP production-and is a key indicator of cell viability. The present study demonstrated that the mitochondrial membrane potential could be a novel biomarker for benzene toxicity in hematopoietic tissues and cell lines. In comparison to nuclear DNA, mtDNA intrinsically exhibits a 10- to 20-fold higher susceptibility to genetic alterations. Therefore, mtDNA is regarded as a useful molecular target to monitor genotoxicity caused by environmental hazards [34].
We identified biomarkers which can indicate the exposure of benzene in blood cells and hematopoietic tissues [34]. The mitochondrial content and membrane potential increased dramatically after three weeks of direct benzene exposure. The hydrogen peroxide level, mtDNA copy number, and numerous protein markers increased after a few weeks of benzene treatment, compared to the group not treated with benzene. However, the heterogeneous nuclear ribonucleoprotein (hnRNP) A2/B1, decreased after exposure to benzene. Thus, increased mitochondrial mass, mtDNA copy number, and the hnRNP A2/B1 protein can be used as biomarkers for benzene-related toxicity and hematotoxicity [34].
Moreover, the mitochondrial proteome was studied to determine whether biomarkers associated with exposure to benzene could be identified. The results indicated that the hnRNP A2/B1 protein may be a novel biomarker associated with benzene exposure. hnRNPs are abundant and exhibited "multi-tasking" features by playing a central role in RNA metabolism; the levels of hnRNPs significantly decreased after exposure to benzene. These proteins are involved in packaging nascent RNA, alternative RNA splicing, mRNA export from the nucleus, and cytoplasm trafficking, stability, and translation. In addition, hnRNPs may play a poorly defined role in telomere maintenance [34].
Our previous work showed that after benzene exposure, there was a decline in hnRNP A2/B1 levels and cell cycle arrest, which led to decreased numbers of primary blood cells and cell lines. hnRNP B1, an RNA binding protein, is overexpressed in early-stage lung cancer, including bronchial dysplasia, a premalignant lesion of lung squamous cell carcinoma. Plasma hnRNP B1 mRNA has been reported to be a useful noninvasive marker for the detection of lung cancer [34].
In summary, in response to elevated intracellular ROS levels, the direct exposure to benzene by primary blood cells and cell lines in vitro was associated with increased mitochondrial mass and higher mtDNA copy number. These results were consistent with the observation that mitochondrial mass and mtDNA contents are increased for the cell to respond to endogenous or exogenous oxidative stress. In addition, results of the present study identified several useful biomarkers associated with benzene exposure, including a novel hnRNP A2/B1 protein.
Polycyclic aromatic hydrocarbons (PAH) are ubiquitous environmental toxicants found in air, water, plants, and soils [35]. Owing to their widespread dispersion in the environment and the adverse health effects associated with PAH exposure (e.g., carcinogenesis and endocrine disruption), there has been increasing concern in the human health field regarding PAH exposure. Although the adverse effects associated with each individual PAHs are not entirely analogous to one another, the United States Environmental Protection Agency has designated 32 PAH compounds as priority pollutants (www.epa.gov). The toxicities of PAHs are determined by their structures. Among the 32 PAH compounds, benzopyrene is notable as the first chemical carcinogen to be discovered [36, 37].
CONCLUSIONS AND FUTURE DIRECTIONS
Mitochondria are important micro-organelles that produce the energy fueling cellular development, differentiation, and growth. Owing to the lack of introns in mtDNA, the rate of genetic mutation is 10- to 20-fold higher in mtDNA compared to nuclear DNA. The mtDNA repair system is inefficient, rendering the mitochondrial genome more susceptible to ROS produced during the ATP synthesis process within the mitochondria. ROS are commonly released during chronic inflammation, and mtDNA is one of the targets for ROS and free radicals. ROS-induced inflammation and excess stress can overwhelm the antioxidant system and lead to mtDNA damage. This oxidative stress plays an important role in mitochondrial dysfunction and mitochondrial genome aberrations, precipitating in various human diseases including hematological malignancies, solid cancers, chronic inflammatory diseases, and diseases caused by environmental hazards.
 XML Download
XML Download