

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
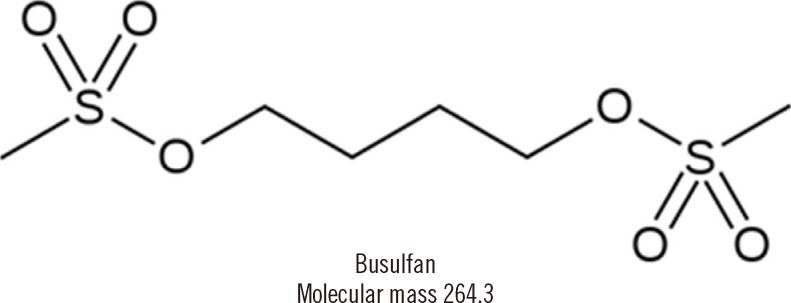
Busulfan (butane-1,4-diyl dimethanesulfonate, Fig. 1) is an alkylating agent, which is commonly used as a component of myeloablative regimens prior to hematopoietic stem cell transplantation (HSCT) [1]. It may be used in association with cyclophosphamide or fludarabine, as an alternative to regimens that involve total body irradiation [2]. Oral administration exhibits wide inter- and intra-individual variability in plasma levels due to vomiting and highly variable bioavailability [3]. In contrast, when administered as intravenous (IV) formulation, pharmacokinetic (PK) features are more predictable through the removal of effect of oral bioavailability and precise administration. For this reason, IV busulfan is gradually replacing oral busulfan but significant inter-individual variability is still observed, especially in children [4, 5].
Therapeutic drug monitoring (TDM) based on area under the curve (AUC) or steady state concentration has been widely investigated [6, 7]. The major purpose of TDM is to prevent drug-related toxicity (for example, hepatic veno-occlusive disease, interstitial pneumonia) while maintaining high-dose busulfan to achieve effective myeloablation prior to HSCT. Inappropriately low drug levels may lead to relapse and even graft rejection, while high plasma busulfan levels are related to high incidence of complications [8, 9]. Currently, most institutes use AUC acquired from serial monitoring of post-administration concentrations to assess PK features in pediatric patients undergoing HSCT. For proper and rapid calculation of AUC, timely and accurate determination of plasma busulfan concentration is crucial.
A number of chromatographic techniques coupled with a few detection methods have been described for analyzing busulfan in plasma and in other biological fluids. Gas chromatography (GC) coupled with electron capture detector [10] or mass spectrometry (MS) [11], liquid chromatography (LC) coupled with UV detectors [12] or fluorescence detectors [13] have been introduced, and an ELISA-based automated method was developed recently [14]. Among the existing methods, LC coupled with MS [15, 16] or with tandem MS (MS/MS) [17, 18] offers a high level of sensitivity and requires a small sample volume, which is beneficial in a pediatric setting. Moreover, other major advantages include the possibility to eliminate complex derivatization procedures and the requirement of only a 10-min run. Recently, modified MS/MS methods employing turbulent flow extraction technology [19], or using dried blood spots as samples [20] were introduced.
We describe a simple, rapid, and sensitive LC-MS/MS assay for accurately quantifying busulfan in human plasma. This method was validated for the parameters of precision, recovery, matrix effect, linearity, detection capability, carryover effect, and stability. In addition, the method was applied to a clinical setting for measuring plasma busulfan concentration in pediatric patients.
Go to : 
METHODS
1. Chemicals and reagents
Glipizide purchased from Sigma Chemical Co. (St. Louis, MO, USA) was used as an internal standard (IS). Two milligrams of glipizide were dissolved into 10 mL of acetonitrile (ACN, Avantor Performance Materials, Center Valley, PA, USA) to make 0.2 mg/mL of IS stock solution. Working IS solution containing 100 ng/mL of glipizide was made by diluting the stock solution with 80% ACN. Busulfan (Sigma Chemical Co.) was dissolved into ACN to make 0.5 mg/mL of stock solution. Samples used for calibration and quality control were prepared by diluting the stock solution with Lyphochek drug-free plasma (Bio-Rad Laboratories, Irvine, CA, USA).
2. Sample preparation
Whole blood was collected in EDTA tubes and was centrifuged at 1,900×g for 10 min. Fifty microliters of the supernatant plasma was mixed with 450 µL of working IS solution, vortexed, and centrifuged at 3,750×g for 10 min at 10℃. One hundred microliters of the supernatant was transferred into injection vials for chromatographic analysis.
3. LC-MS/MS analysis
Agilent 1260 Infinity (Agilent Technologies, Santa Clara, CA, USA) LC system equipped with an XBridge™ C18 column (Waters Co., Milford, MA, USA, 2.1×100 mm, 3.5 µm) was used and eluted using a gradient. The mobile phases were comprised of 0.1% formic acid with 2 mM ammonium acetate in distilled water (solvent A) or methanol (solvent B). Following sample injection (50 µL), elution was performed by generating a gradient from 10 to 50% solvent B in the starting/initial 2 min and 30 sec, followed by a run with 50% solvent B for 4 min, then rapidly returned to the initial conditions and stabilized for 3 min. The total run time was 10 min per run and the flow rate was maintained at 0.35 mL/min at 40℃. The retention time was 2.27 min for busulfan and 4.89 min for IS (Fig. 2).
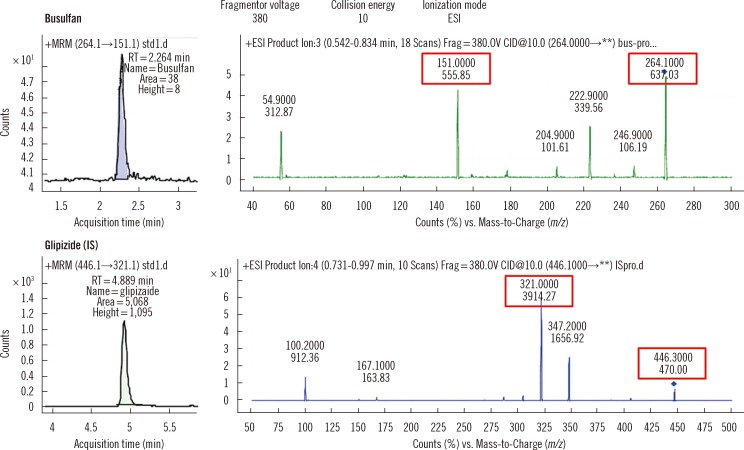 | Fig. 2Mass spectra and chromatograms of busulfan and internal standard obtained from the analysis of the lowest calibration solution (25 ng/mL). The MRM chromatograms (left panels) show well-separated busulfan and glipizide as indicated by different retention times. The right panels show the MRM transitions of busulfan and glipizide.
Abbreviations: IS, internal standard; MRM, multiple reaction monitoring; ESI, electrospray ionization.
|
The LC system was coupled with triple quadrupole MS/MS, Agilent 6490 (Agilent Technologies). The MS/MS was equipped with an electrospray ionization (ESI) source, operated in the positive ion mode, and the quantification was performed in the multiple reaction monitoring (MRM) mode with mass-to-charge (m/z) transitions at 264.1>151.1 for busulfan and 446.1>321.1 for IS (Fig. 2). Nitrogen gas was used for nebulization, desolvation, and collision. The instrument conditions were set as follows: collision energy 4 V for busulfan and 8 V for IS, capillary voltage 4.0 kV, dwell time 200 ms, sheath gas flow 12 L/min at 300℃, desolvation gas flow 14 L/min at 250℃, and 50 psi nebulization gas pressure. The injection volume was 2 µL and the data was acquired and analyzed using MassHunter Workstation software (Agilent technologies). The ratios of the peak areas of busulfan to that of IS were used for all calculations.
4. Preparation of calibrators and quality control (QC) samples
The samples used for calibration and QC were prepared by mixing busulfan stock solutions with drug-free plasma as recommended in the Food and Drug Administration (FDA) guidance for industry [21]. Six different concentrations of calibration solutions were prepared for obtaining a 6-point standard curve (25, 50, 200, 500, 2,000, 5,000 ng/mL) and 3 concentrations of samples were prepared for QC (200, 1,000, 4,000 ng/mL). The concentrations of the calibration solutions and QC samples were selected to cover the targeted busulfan plasma levels measured during routine monitoring. A calibration curve was generated using y=ax+b applying the quadratic regression weighed by 1/x where x is the spiked concentration of busulfan and y is the ratio of the peak area of busulfan against that of IS.
5. Validation procedure
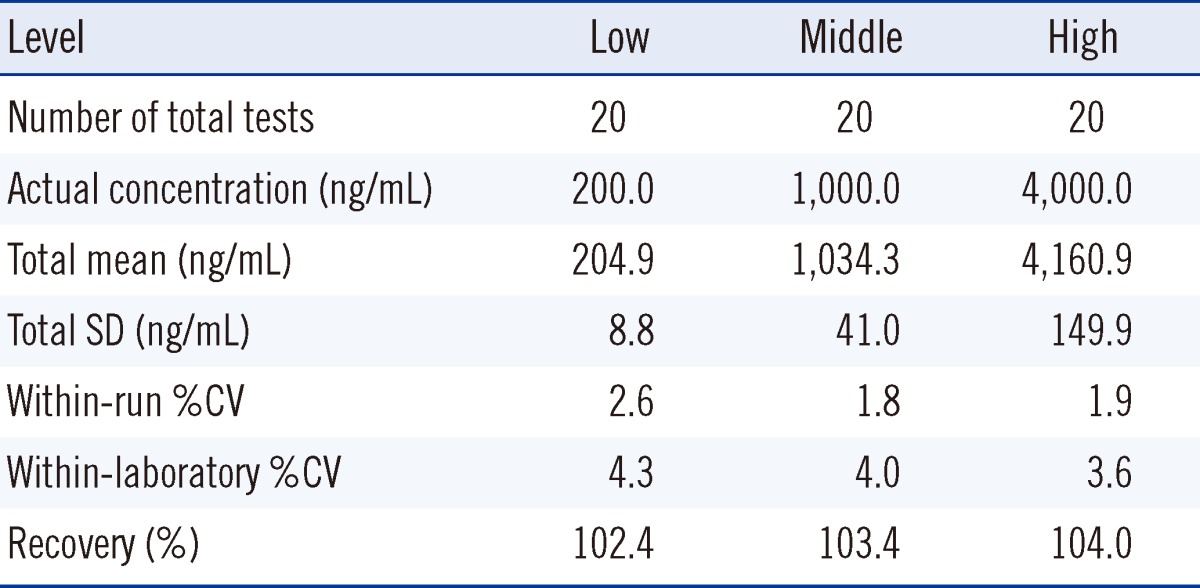
Within-run and within-laboratory precision were evaluated according to the procedures described in the CLSI guideline EP15-A2 [22]. The precision study was performed by assaying 3 levels of QC samples for 5 consecutive days in quadruplicates per day, overall 20 replicates per a concentration. In this procedure, recovery of analytes also could be calculated, because QCs were made by spiking of certain amount of purified busulfan into drug-free plasma.
For evaluation of the matrix effect, plasma samples without busulfan were prepared in the same way as patient samples were. An equal amounts of busulfan and IS were spiked into the prepared plasma samples and the stock solutions. The busulfan concentrations were analyzed and the matrix effect was calculated as the ratio of the concentrations in the stock solutions and the prepared plasma samples [23].
Linearity was evaluated according to the procedures described in CLSI guideline EP6-A [24]. Samples with 5 levels of busulfan (range, 0-5,000 ng/mL) were prepared by mixing drug-free plasma and busulfan-spiked plasma at varying ratios, assayed in quadruplicates, and the mean value was used to assess the linearity of the method.
The limit of detection (LOD) was determined as the lowest concentration with signal to noise ratio higher than 3 [25]. The limit of quantification (LOQ) was set as the lowest concentration of the calibration solution at CV less than 20% and recovery within 80-120% [21]. Two different QC samples (200 and 4,000 ng/mL) were used to investigate the carryover effect, and the order of analysis was as follows: high-high-high-high-low-low-low-low. Each result was designated separately as H1, H2, H3, H4, L1, L2, L3, L4, and the carryover was calculated by the equation: carryover %= 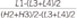 ×100 as previously described [26].
×100 as previously described [26].
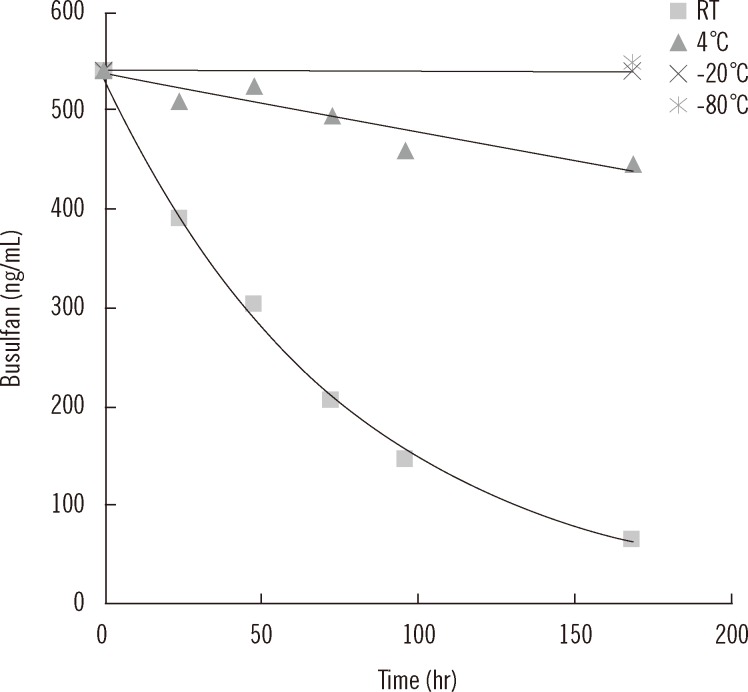
×100 as previously described [26].The short-term stability of busulfan in plasma was evaluated at various storage conditions. EDTA samples were spiked with 540 ng/mL busulfan and stored at room temperature and at 4℃ for 1 week and at -20℃ and -80℃ for 2 weeks. Plasma samples stored at room temperature and at 4℃ were analyzed on days 1, 2, 3, 4, and 7, and samples frozen at -20℃ and -80℃ were analyzed after 1 week and 2 weeks of storage, respectively. The validation data was analyzed using Microsoft Excel 2010 (Redmond, WA, USA).
6. Clinical samples
Peripheral blood was collected in EDTA tubes from 9 children. IV busulfan was administered to the patients once daily for 4 consecutive days before HSCT [5]. On each day, samples were collected just after the administration and subsequently after 1,2, and 4 hr. The dosage of busulfan on the 1st day of the schedule was 120 mg/m2 for children over 1 yr of age and 80 mg/m2 for infants below 1 yr of age. From day 2, the busulfan dosage was adjusted based on the previous day's TDM results. The distribution of the busulfan concentrations was plotted according to the box-whisker plot using MedCalc version 12.4 (Ostend, Belgium).
Go to :
RESULTS
The mass spectra and chromatograms of busulfan and IS acquired from the analysis of the calibration solution containing the lowest busulfan concentration (25 ng/mL) are shown in Fig. 2. The spectra showed no interfering peaks around the retention time of busulfan and IS from the calibration solutions, QC samples, and clinical samples.
1. Precision, recovery, and matrix effect
The precision and recovery of the method were all within the acceptable range on using QC samples prepared by mixing a large amount of drug-free plasma with busulfan solution of known concentration (Table 1). The within-run and within-laboratory CVs were all below 5%, which were acceptable for clinical use. Recovery of busulfan was evaluated by comparison of the measured value with the expected value in busulfan-spiked plasma. The % recovery was between 100% and 105%, which was acceptable for clinical uses. The matrix effect was 100.3-107.5% in the range of 220-2,887 ng/mL.
2. Linearity, detection capability, and carryover
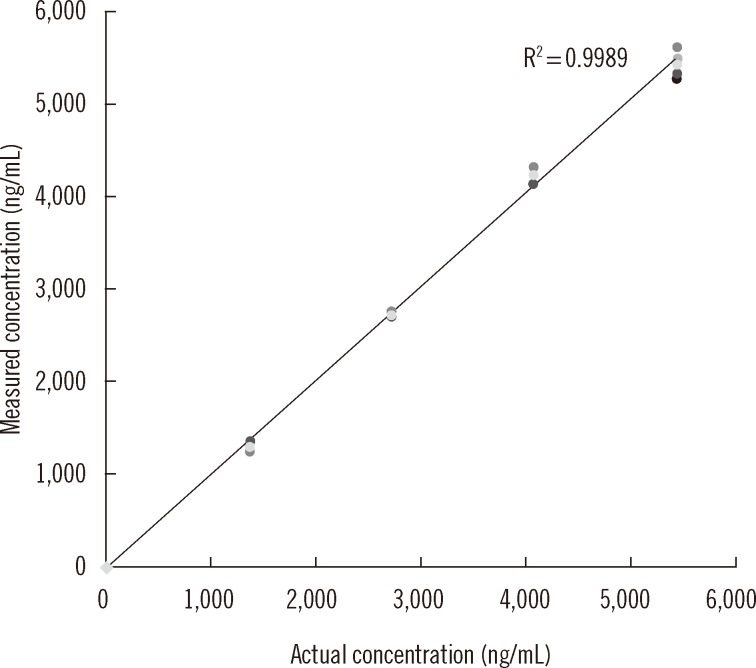
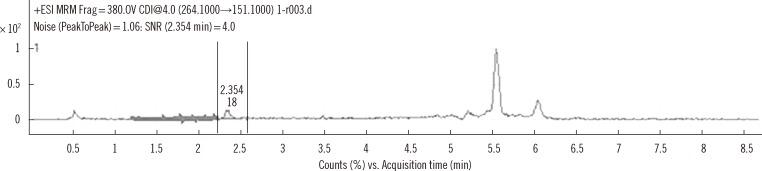
In Fig. 3, the plot displayed linearity from 0 to 5,000 ng/mL, which covered the range of busulfan concentrations from most of clinical samples. Busulfan was not detected in any of the 20 samples from patients that had not been administered busulfan, confirming specificity of the assay for detection of busulfan in plasma. The LOD was 1.56 ng/mL, at which, the signal to noise ratio was 4.0 (Fig. 4). The LOQ was set as 25 ng/mL, the lowest concentration of the calibration solution that displayed a CV of 7.4% and a recovery of 95.2%. The carryover was under 1%, which was within allowable limits for clinical tests.
3. Stability of busulfan in plasma
The busulfan concentration decreased significantly especially on being stored at room temperature, and 24 hr after storage, the concentration decreased by 25%, in comparison to less than 5% at 4℃. On storing for 2 weeks at -20℃ and -80℃, the change in busulfan concentration was negligible (Fig. 5).
4. Clinical samples
The analysis results ranged 347-5,076 ng/mL, and the concentrations were primarily dependent on the elapsed time post administration as expected. The median (interquartile range) busulfan concentration in plasma at 0, 1, 2, and 4 hr after the 1st administrations were 3,999 (3,770-4,152), 3,281 (2,673-3,579), 2,340 (1,979-2,708), and 1,249 ng/mL (893-1,558 ng/mL), respectively (Fig. 6).
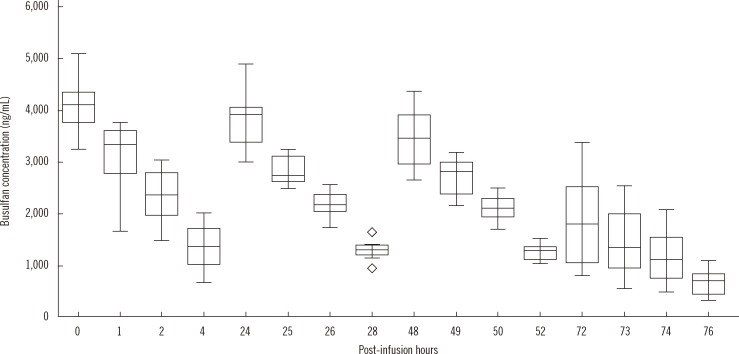 | Fig. 6Distribution and change of plasma busulfan concentrations in clinical samples. The central box represents the values from the lower to upper quartile (25-75 percentiles). The middle line represents the median value. A line extends from the minimum to the maximum value, excluding "outside" and "far out" values displayed as separate points (marked as ⋄).
|
Go to :
DISCUSSION
We describe a simple and rapid method for the quantification of plasma busulfan using LC-MS/MS. LC-MS/MS is the most preferred method for measuring busulfan in a pediatric setting because of its benefits over other methods. GC-based methods and LC-fluorescence/UV techniques are time-consuming and laborious, requiring derivatization steps for preparation and a large quantity of plasma of up to 1 mL, along with a long analysis time of up to 30 min per run [10-13]. The recently developed ELISA-based automated method shows similar precision, required time, and sample volume compared with previously described methods, while having the risk of cross-reactivity and lot-to-lot variability of reagents [14]. In contrast, the LC-MS/MS provides the highest sensitivity and specificity, minimizes the preparation steps, and requires the smallest sample volume along with remarkably short analysis time [17, 18, 27].
In our method, plasma samples were pretreated with very simple liquid-to-liquid extraction procedure before analysis requiring only about 10 min, followed by the chromatographic analysis that required 10 min for 1 run. Short run time enables rapid reporting of busulfan concentrations to the clinicians, which helps them decide whether to adjust next day's dosage. Moreover, this assay requires only 50 µL of plasma, which can be advantageous for infants and children. Such characteristics could be attractive for institutes, which are considering or planning to implement TDM of busulfan for pediatricians.
In comparison with other known LC-MS/MS methods [17, 18, 27], a run time of 10 min required for chromatographic analysis is somewhat longer than the 3-4 min run time in previously described methods. However, considering the time needed for sample preparation, the total elapsed time is nearly as much as or slightly shorter than previous methods, as the present method does not require complex extraction procedures or evaporation processes. Besides, this difference may be attributed to the temporal gradient of the mobile phase and time required for stabilization of the chromatographic column. The amount of sample required for this method (50 µL) was slightly less than the previously known methods (50 to 200 µL). Further, all known LC-MS/MS methods employed ESI and MRM in positive ion mode, and there are subtle differences in the use of IS, conditions for chromatographic and mass spectrometric analysis.
Studies on precision, recovery, matrix effect, and linearity showed acceptable results. Compared with earlier studies, our method showed similar performances in precision, recovery, and matrix effect. However, this study was validated to be linear over the entire range of 2,000-5,000 ng/mL, which had not been fully validated in previous studies. This could be due to the difference in the interval and dosage of busulfan in each facility. In our hospital, samples with higher busulfan concentrations than 2,000 ng/mL were frequently observed. Thus, the linearity of the test had to be validated including these ranges. On the other hand, relatively low concentrations (<500 ng/mL) were not frequently observed in our clinical evaluation process, which makes verification of precision in lower range less important than in the previous studies, so precision tests with low QC (200 ng/mL) are enough for medical uses.
Implementation to the clinical setting was largely successful, but was accompanied with a problem in assaying samples collected during weekends. Busulfan was found to be unstable in human plasma, especially at room temperature, which is similar to previous report [15], and it was assumed that busulfan may react with nucleophilic groups such as amines in plasma during storage. Fortunately, the reduction in concentration of busulfan occurs at lesser extent on storing the samples in a refrigerator. Therefore, in laboratories planning to establish analysis of plasma busulfan, it is essential not to store samples at room temperature before analysis.
There are some limitations in our method. We have not compared the results with those of other methods or institutes, because there are not adequate laboratories or accounts on the measurement of busulfan in plasma in clinical laboratories in Korea. To our knowledge, widely used external proficiency testing programs do not provide the programs for inter-laboratory comparison. Recently, some reports on determination of metabolism of busulfan by analyzing urine or plasma [28-31] have appeared, and we have also searched for candidate metabolites that could be used to assess individual variability in metabolism. Since the relationship with busulfan-related toxicity and concentration of specific metabolites is not yet clarified, studies for metabolites can be candidates for further evaluation.
In summary, we have developed an accurate and reproducible method for quantification of busulfan in human plasma using LC-MS/MS. The method requires relatively short analysis time and simple preparation procedures. Thus, this can be a useful protocol for laboratories especially in pediatric settings and considered for introduction of TDM of busulfan to achieve safe and proper dosing.
Go to :
 XML Download
XML Download