

 PDF
PDF ePub
ePub Citation
Citation Print
Print


According to the International Organization for Standardization (ISO) 14971, risk management is described as the systematic application of management policies, procedures and practices to the tasks of analyzing, evaluating, controlling, and monitoring risk [1]. It is a process that involves anticipating what could go wrong (errors), assessing the frequency of occurrence of these errors, as well as the consequences or severity of harm they cause and finally what can be done to reduce the risk of potential harm to an acceptable level. The practice of risk management is one that is regularly employed in aerospace and automotive industries, where manufactured products are put through rigorous risk assessments before they are made available to the public. Similarly, in vitro diagnostic device (IVD) manufacturers follow risk management protocols to determine the best use of their devices and then outline the limitations and interferences that can affect the devices in package inserts or user manuals. Risk management, however, is a new concept for clinical laboratories. Because a majority of the standards and guidelines on risk management are geared toward manufacturers, few resources on risk management exist for clinical laboratories. Nevertheless, medical directors can borrow from the industrial principles of risk management to reduce errors in the clinical laboratory. A few recent guidelines, such as the CLSI document EP18-A2, "Risk management techniques to identify and control error sources" [2] or the CLSI guideline EP23-A, "Laboratory quality control based on risk management" [3], introduce risk management to the clinical laboratory. The purpose of this review is to highlight how risk management principles can be utilized in the clinical laboratory to prevent medical errors and minimize harm to patients.
RISK DEFINITION
Laboratory testing of patient samples is a complex process. Errors can occur at any point in the testing process. So, laboratories must take steps to ensure reliable and accurate results are produced. The laboratory must examine its processes for weaknesses or hazards where errors could occur and take action to detect and prevent errors before they affect test results. This can be done by mapping the testing process or following a sample through the preanalytical, analytical and postanalytical stages of testing and examining each step in the process for risk of potential hazards. Risk is defined as the chance of suffering or encountering harm or loss. Risk can be estimated through a combination of the probability of occurrence of harm and the severity of that harm [4]. There is a spectrum of risk from very low to very high risk, and one can never achieve zero risk. Events that occur more frequently pose greater risk and events that cause greater harm are higher risk. Thus, our role as laboratory directors is to manage risk in the laboratory to a clinically acceptable level, a level that is acceptable to our physicians, our patients, and our administration. Risk is essentially the probability of an error occurring in the laboratory that could lead to harm. Harm can occur to a patient, but may also be assumed by the technologist, the laboratory director, the physician, and even the hospital organization as a consequence of a laboratory error. Risk can also be estimated through detectability, which is intended to detect and prevent errors before they leave the laboratory and touch a patient. The analysis of quality control is one example of a detection mechanism employed by laboratories to alert technologists to test system errors before they impact patient results.
RISK ANALYSIS METHODS
Risk analysis can be divided into two main parts, as described in CLSI document EP18. The first part involves a failure modes and effects analysis (FMEA), which identifies potential sources of failure and determines how such failures affect the system. Specifically, a FMEA involves discovering possible sources of failure, determining the probability and consequences of each failure, and outlining control measures to detect and eliminate such failures. For this reason, FMEA is considered a bottom-up approach. Manufacturers will typically perform a FMEA when a process, product or service is being designed or when an existing process or product is being applied in a new way. It is the responsibility of the manufacturer to disclose all sources of failure to the consumer, along with recommendations on how to control and manage these failures. In the clinical laboratory, a FMEA should be conducted before a new assay or instrument system is put in place. The laboratory should consult the manufacturer product inserts to determine already identified hazards, and then pinpoint potential laboratory-specific hazards at the different process steps and outline control measures to prevent these failures. A similar technique to review probable sources of failure is the fault tree analysis (FTA). FTA is a top-down approach that begins by assuming a high level hazard and then determining the root cause of the hazard. It is useful when examining multiple failures and their effects at a system level. A FMEA and FTA should be conducted together to fully assess all possible ways a laboratory system can fail and how to reduce the occurrence of failure.
The second part of the risk analysis process entails reducing the rate of observed failures through a failure reporting and corrective action system (FRACAS). A FRACAS details the failures that have occurred in a system and the control measures employed to correct these failures. The control measures can describe how to prevent the actual failure from occurring again or how to prevent the downstream effects of the failure. A FRACAS is carried out by manufacturers after they design a product but before the product is released to the user, and is essential to understanding how a device or system actually performs from a reliability and maintenance standpoint. The clinical laboratory should perform a FRACAS on all existing laboratory processes to correct the causes of observed errors.
LABORATORY RISK MANAGEMENT
Although risk management techniques and standards have traditionally been targeted to manufacturers, new guidelines such as CLSI document EP23-A [3] are evolving to introduce risk management principles to the clinical laboratory. This document is directed toward laboratory staff and outlines how to develop and maintain a quality control plan (QCP) for medical laboratory testing based on industrial risk management principles. The implementation of EP23-A should not be difficult for laboratories since they already perform activities that could be considered risk management, including assessing the performance of new instruments and assays before testing patient samples, performing regular maintenance and quality control (QC), responding to physician complaints, and troubleshooting errors. The QCP identifies weaknesses in the preanalytical, analytical and postanalytical phases of testing and delineates specific actions to detect, prevent and control errors that can result in patient harm.
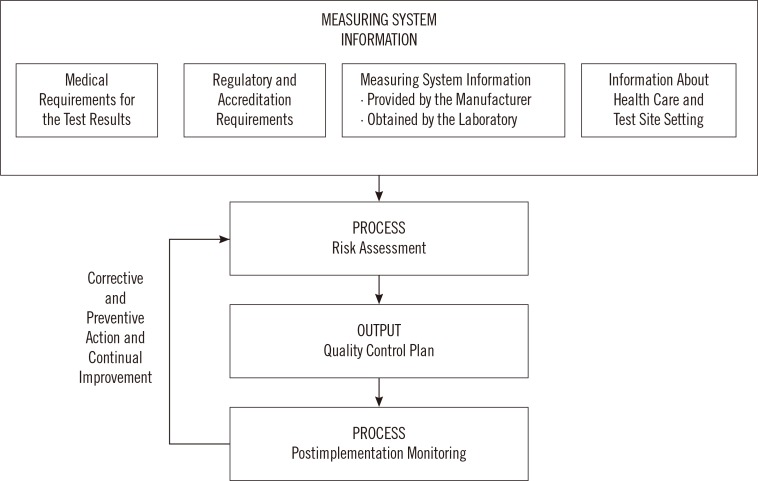
The development of a QCP can be divided into four steps.(Fig. 1) The first step involves collecting system information, including manufacturer recommendations on the proper use of devices or assays, the medical application of test results (how test results influence patient management, are test results used for screening versus diagnosis) as this will define performance specifications and allowable tolerance limits for error, and applicable regulatory and accreditation requirements. The laboratory should also determine how conditions unique to the laboratory, including testing personnel and environmental conditions, can impact risk and the probability of error. Next, the laboratory carries out a risk assessment and identifies control measures to mitigate the potential for harm. The third step summarizes the quality control plan as a list of hazards identified and actions the laboratory must take to minimize risk. Lastly, the quality control plan is implemented and monitored for effectiveness. If errors are noted, then corrective and preventive action (CAPA) is taken to modify and improve the QCP. So, the initial risk assessment and quality control plan is continuously improved over time to ensure that all known risks are well controlled, and no new hazards are identified.
RISK ASSESSMENT
No laboratory test or process is without risk. Moreover, because the laboratory testing process involves numerous steps, the number of potential errors can be large. It is therefore important to assess and prioritize risks and determine what level of risk is acceptable in the clinical laboratory. A FMEA is performed to identify weaknesses, determine the probability and severity of harm that could arise from errors in weak steps of the testing process, and describe controls to detect and prevent such errors. This is best done through process mapping. Each stage of testing is examined to identify possible failure points and control processes that can be implemented to detect and prevent errors. All constituents of the measuring system, starting with the patient sample, reagents, environmental conditions that could affect the analyzer, the analyzer itself, and the testing personnel, are considered in the evaluation of possible failures. An estimate of the occurrence of these failures, whether frequent, occasional or remote, as well as the likelihood of harm arising from each failure is determined. The combination of frequency and severity of harm allows the laboratory to estimate the criticality or risk of the error. Criticality allows the laboratory to address high risk failure modes first and determine the clinical acceptability of low risk events. For example, a grossly hemolyzed sample can lead to an elevated potassium level. If hemolysis is not recognized in the patient's medical record, the clinician could misinterpret the elevated potassium leading to patient harm from inappropriate treatment. Errors that involve incorrect or delayed patient results that affect medical decisions are generally considered more severe than errors that lead to no change or confirmatory follow-up prior to patient treatment. The degree of harm is defined using a semiquantitative scale of severity levels, ranging from negligible harm causing inconvenience or temporary discomfort, to critical or catastrophic harm causing permanent impairment or patient death [3].
RISK CONTROL
After evaluating possible failure modes in the testing process and estimating their criticality or risk, the laboratory then selects appropriate control measures to detect or prevent the error from reaching the patient, and maintain risk at a clinically acceptable level. QC is intended to monitor the performance of a measuring system and inform when failures arise that could limit the usefulness of a test result for its intended clinical purpose. A common way to monitor the stability of an analytical system is through the use of liquid QC material. The laboratory establishes ranges and control rules that define how much change in assay performance is allowed before the QC results are considered out-of-control. Westgard rules are one such example that employ limits calculated from the mean value and standard deviation of control samples measured when the system is stable, and describe when to accept or reject QC results [5]. The use of multiple control rules can improve error detection while maintaining a low likelihood of false rejection.
QC samples should be tested in the same manner as patient samples and must be run on a frequent basis. In general, a minimum of two levels of QC each day of testing is recommended. However, the frequency of QC testing should reflect the test system risk and be based on the stability of the analyte and the measuring system, the presence of built-in controls, the number of patient samples processed, the clinical use of the test results and the frequency of calibration [6]. The frequency of QC testing must also conform to regulatory and accreditation requirements. Measurement of QC samples is useful in detecting systematic errors that affect all test results in a predictable manner. For example, liquid QC is very effective at detecting errors caused by faulty operator technique (pipette or dilutional errors) or incorrect reagent preparation that affect both patient and QC samples in the same manner. However, liquid QC does not address all potential failure modes. Random and unpredictable errors such as hemolysis or lipemia that affect individual samples are poorly detected by liquid QC. Preanalytical errors that occur prior to the sample arriving in the clinical laboratory (specimen mislabeling) and postanalytical errors such as incorrect result entry are also not detected by liquid QC.
For this reason, many test systems employ alternative QC strategies, in addition to liquid QC, to ensure that the potential for high risk errors is covered. Newer laboratory instruments and point-of-care testing devices incorporate a variety of biologic and chemical controls and system electronic checks engineered into the test system to address a number of potential errors. Bubbles and clot detection can sense problems with specimen quality and alert the system operator with an error code rather than a numerical test result. Unit-use point of care tests, like urine pregnancy or occult blood cards are consumed in the process of analyzing a control sample. So, liquid QC provides little assurance that the next test will perform in the same manner. Such tests include built-in control lines or areas that can detect incorrect test performance and test mishandling or storage degradation with each test. Molecular lab-on-a-chip tests perform 100 or more reactions on the same cartridge. Analysis of two levels of liquid QC on each reaction each day of testing is neither practical nor possible. Therefore, laboratories need to employ other control processes that address the risk of errors that are most likely to affect test results such as the quality of the specimen, the reactivity of the polymerase enzyme, and the temperature cycling of the instrument. Other devices, like transcutaneous bilirubin are noninvasive and cannot even accept a blood or QC sample. Thus, alternative control processes must be utilized to ensure the quality of testing with such devices.
Laboratories have a variety of control processes at their discretion. Patient samples can be used as their own controls through calculation of running averages of test results to indicate drift or shift in analyzer performance over time. Manufacturers have started encoding expiration dates within reagent barcodes to prevent use past the expiration date. Modern automated analyzers can also be programmed to detect specimen errors such as hemolysis, lipemia and icterus, and warn physicians of potential interferences that would affect the interpretation of test results. Laboratory information systems can be designed to flag and hold test results that appear physiologically improbable or exceed predefined limits. These unbelievable results can then be reviewed against previous patient results by the instrument operator to ensure their validity. Similarly, delta checks that detect significant differences between the current and previous test result for the same patient can flag and hold the result for operator review before release. Delta checks are particularly useful in detecting preanalytical errors such as mislabeled samples.
External quality assessment or proficiency testing (PT) is another control process the laboratory can use to ensure test system performance. Samples are mailed periodically to the laboratory from an external quality assurance program, such as the College of American Pathologists (CAP). These samples are analyzed like patient samples and results are returned for grading by comparison to the results of other laboratories using the same make and manufacturer of laboratory instrumentation. If a laboratory's results exceed the total allowable error (which accounts for imprecision and bias), the laboratory's test system performance may have drifted from their peers, signaling a failure that may require troubleshooting.
MONITORING THE QUALITY CONTROL PLAN
Once all of the weaknesses in the testing process have been identified and appropriate control processes selected to address each weakness, these hazards and control processes are summarized as a QCP. The QCP is implemented by the laboratory for that test and monitored for effectiveness to ensure that errors are adequately being detected and prevented. A QCP can be monitored by reviewing quality benchmarks for the test such as physician complaints. When a complaint is received, the laboratory should troubleshoot to determine what occurred and how to prevent recurrence of the error in the future. The QCP should be reassessed to determine if this is a new failure not considered during development of the initial QCP, or whether this is a hazard occurring at higher frequency or with greater severity of harm than previously considered. Once risk is reassessed, the QCP should be appropriately modified to maintain risk to a clinically acceptable level, and the modified QCP implemented. The laboratory should also remain informed of any circumstances that could impact the QCP, including manufacturer recalls or product updates, and adopt the QCP to these changes. For example, if an error occurs after implementation of a QCP that is due to incorrect temperature monitoring of a refrigerator that stores reagent, the laboratory should institute new control processes to prevent the error from recurring in the future. The laboratory can reeducate staff on the impact of improper reagent storage on assay performance and patient results, reinforce daily temperature checks and ensure that each refrigerator is outfitted with a continuously monitored thermometer or even an alarm system to alert when the temperature drifts out of the expected range. Furthermore, each refrigerator can be placed on a backup generator such that in the case of a power outage, the refrigerator maintains the correct temperature and reagents are not affected. These changes are then incorporated into the QCP, and the effectiveness of the new control processes monitored periodically to make certain the corrective actions are working.
CONCLUSION
Risk management is a practice developed in industrial or manufacturer settings. However, new guidelines have been published to introduce risk management principles to the clinical laboratory. Risk management can minimize the chance of errors and ensure reliability of test results. Risk management guidelines recommend that laboratories play a proactive role in minimizing the potential for errors by developing individualized QCPs to address the specific risks encountered with laboratory analysis. Laboratories should map their testing process to identify weaknesses at each testing step. As risks are identified, the laboratory selects appropriate control processes to detect and prevent errors from occurring. All the hazards and control processes are summarized in a QCP. Once implemented, the efficacy of the laboratory's QCP should be continuously monitored and revised as further errors are identified, to ensure that patient results are reliable and residual risks are maintained to a clinically acceptable level.
 XML Download
XML Download