

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatitis E virus (HEV) causes acute, self-limited hepatitis [1, 2] and is endemic in many developing countries, especially in South and Southwest Asia, North Africa, and the Middle East [3]. HEV belongs to the Hepeviridae family [4]. Its genome consists of a positive single-stranded RNA containing 3 overlapping open reading frames (ORFs), ORF1, ORF2, and ORF3 [5, 6]. According to the phylogenetic analysis, HEV has 5 genotypes with different geographical distributions and hosts [7]. Humans are most often infected with genotypes 1 and 2, while pigs and other animals are most often infected with genotypes 3 and 4 [8]. Genotype 5 has been identified in avian species and is non-infectious for humans [4]. Genotype 1 predominates in Asia and North Africa [9]. HEV is transmitted via the fecal-oral route, usually from drinking water [1]. According to the epidemiological data, one-third of the world's population is infected with HEV [2]. The mortality rate has been estimated to be 1-15% in the general population [7]. The acute infection is self-limited in healthy individuals and does not become chronic; however, HEV may cause serious consequences in pregnant women and patients with chronic liver disease [2, 7]. The infection is life-threatening during pregnancy, causing fatal and fulminant hepatitis with a high rate of miscarriage and premature birth [7]. Epidemiological studies have reported a mortality rate of up to 30% in pregnant women [7].
Consequently, HEV is a serious public-health threat, and rapid diagnosis is essential. HEV viremia is limited to the acute phase of the infection; therefore, diagnosis depends on serological assays [10]. Unfortunately, sufficient amounts of natural viral proteins are not available for serological diagnosis, because there are no appropriate culture systems for HEV. Therefore, production of recombinant proteins is required to overcome this problem. Such proteins can then be used for specific antibody detection [10, 11].
Among the HEV genes, ORF1 encodes non-structural proteins, which are not targeted by antibodies [12]. Antibodies formed against the protein encoded by ORF3 are transient, making ORF3 an unsuitable antigen for serological diagnosis of HEV [13]. ORF2 encodes a capsid protein of 72kDa (660 amino acids). It is suitable for serological diagnosis of HEV and is a candidate for a vaccine against HEV infection, because it is immune-dominant and highly conserved among HEV species, and induces long-lived immunity [9, 13, 14]. When expressed, the full-length capsid protein (72 kDa) is not a suitable diagnostic target, because the important epitopes are relatively hydrophobic, insoluble, and therefore masked. However, truncated forms of the capsid protein are considered diagnostic antigens. Among several truncated forms of the full-length ORF2 protein, the 56-kDa form is more stable and is highly active in the detection of HEV antibodies [15, 16].
Since HEV grows poorly in cell culture, the ORF2 gene or fragments thereof have been cloned and expressed in different expression systems, such as prokaryotes, insect cells, animal cells, and transgenic plants [6]. Recently, different recombinant antigens have been used in the design of assays for diagnosing HEV; these assays have optimized the sensitivity and specificity of the antigens in order to provide the best diagnostic test. In addition to antigen sensitivity and specificity, economical large-scale production of the target proteins is an important goal [11]. Satisfactory expression of ORF2 proteins can be achieved in bacterial and animal cells, but proteins produced in animal cells are not cost effective. Therefore, an economical method of high-yield production of ORF2 proteins is expression in a prokaryotic expression vector system in E. coli cells [6].
In the present study, we described the simple and low-cost development of 2 ELISAs using 2 truncated forms of the HEV ORF2 proteins. We then evaluated the ability of these ELISAs to detect anti-HEV IgG in serum samples and compared our results to those obtained with DIA.PRO HEV IgG ELISA kit (DIA.PRO, Milan, Italy).
METHODS
1. Gene optimization and synthesis
The nucleotide sequence of the truncated ORF2 gene (ORF2.1, encoding amino acids 112-660) of HEV genotype 1, isolate sar55 (Gen-Bank accession number AF444002.1) was analyzed using GenScript Rare Codon Analysis Tool (GenScript USA Inc., Piscataway, NJ, USA). The sequence data was submitted to GenScript, and the optimal gene was designed using GenScript's OptimumGene Gene Design tool (GenScript USA Inc.) for expression in E. coli. To subclone it into the pET30a (+) vector, 2 restriction-enzyme digestion sites, NdeI and XhoI, were placed in the codon-optimized gene. The pET system is one of the best tools for the cloning and expression of recombinant proteins in E. coli BL21 cells, and was, therefore, used for subcloning [17]. Another truncated form of the ORF2 gene was constructed (ORF2.2, encoding amino acids 112-608) from the previously truncated form by using 2 digestion sites for NheI; the first was at amino acid 608, and the second was added after the stop codon. After the second NheI digestion site, an 8-His tag and 2 stop codons were added. To confirm our design, in silico digestion was performed using Clone Manager Basic software version 9 (Sci-Ed Software, Cary, NC, USA), and the translated protein sequences were aligned using sar55 strain by MEGA software version 4.0 (Biodesign Institute, Tempe, AZ, USA) [18]. The optimized coding sequence was then synthesized and cloned into the commercial cloning vector, pBluescript II SK (+) by Biomatik Company (Biomatik Corporation, Cambridge, Canada).
2. Subcloning and construction of the expression plasmid
The pBluescript II SK (+) vector carrying the optimized ORF2.1 gene (pBluescript II SK-ORF2.1) was digested by NdeI and XhoI restriction enzymes (New England BioLab, Ipswich, MA, USA). The expression vector pET-30a (+) (Novagen, Madison, WI, USA) was also digested by the same enzymes in order to subclone the optimized gene. After thermal inactivation of NdeI and XhoI and analysis by agarose gel electrophoresis, the linearized plasmid and the ORF2.1 gene were extracted from the agarose gel by using an Agarose Gel DNA Extraction Kit (Roche, Mannheim, Germany) and used for ligation by T4 DNA ligase (New England BioLab). After ligation, the first recombinant plasmid pET30a-ORF2.1 was generated and transformed into E. coli DH5α competent cells by electroporation as described previously [19]. Transformed E. coli cells were selected on Luria-Bertani (LB) medium (HiMedia, Mumbai, India) agar plates containing kanamycin (50 mg/L). Several colonies were assayed by colony PCR using universal primers for the T7 promoter and T7 terminator. After selecting recombinant clones, the plasmid DNA was extracted from cells cultured overnight by using the High Pure Plasmid Isolation Kit (Roche). The second recombinant plasmid, pET30a-ORF2.2, was obtained by deleting a 193-nucleotide fragment from the first recombinant plasmid by digestion with NheI. The fragment was then ligated using T4 DNA ligase. Finally, the plasmid DNA was extracted and confirmed by PCR, restriction-enzyme digestion, and DNA sequencing. In order to enhance protein expression, the recombinant plasmids were each transformed into competent E. coli BL21 (DE3) cells separately.
3. Expression and purification of recombinant proteins
E. coli BL21 (DE3) cells containing the first recombinant plasmid pET30a-truncated ORF2.1 were cultured overnight, inoculated in Terrific Broth (HiMedia) supplemented with kanamycin (50 mg/L) in a 1:100 volumetric ratio, and grown with shaking at 250 rpm at 37℃ until the optical density at 600 nm (OD600) reached 0.5. Protein expression was induced by adding various concentrations (0.1-1 mM) of isopropyl-β-D-thiogalactopyranoside (IPTG) to the bacterial culture, and the cells were incubated with shaking for different induction times (2, 4, 6, 8, and 16 hr) at different induction temperatures (37℃, 30℃, and 25℃) to optimize the protein expression.
The optimal conditions were determined and large-scale protein expression studies were carried out. Purification by Ni2+-chelate-affinity chromatography (Qiagen, Hilden, Germany) under denaturing conditions was carried out according to the manufacturer's protocol. The induced cells from a 200 mL culture were harvested by centrifugation at 4,000 rpm at 4℃ for 20 min. The cell pellet was suspended in 10 mL of phosphate-buffered saline (PBS) and then lysed by 3 cycles of freeze-thawing in liquid nitrogen and cold water (4℃) and sonication 3 times in 10 sec burst. The lysate was centrifuged at 15,000 rpm at 4℃ for 30 min. To solubilize the inclusion body proteins, the pellet was resuspended in lysis buffer (6 M guanidine HCl, 20 mM NaH2PO4, 500 mM NaCl, pH 8.0) and stirred for 30 min at room temperature. The suspension was cleared by centrifugation at 15,000 rpm for 30 min at 4℃. One milliliter of Ni-NTA agarose (Qiagen) was equilibrated with the lysis buffer and added to the clear supernatant. The agarose-sample suspension was gently shaken at room temperature for 30 min to allow the protein to bind to the agarose, and then centrifuged at 1,000 rpm for 2 min. The supernatant was removed, and the agarose was sequentially washed 3 times with 10 volumes of binding buffer (8 M urea, 20 mM NaH2PO4, 500 mM NaCl, pH 8.0). The agarose was then transferred to a column and sequentially washed twice with 3 volumes of wash buffer (binding buffer with a linear pH gradient of 8.0, 6.5, and 6.0). The protein bound to Ni-NTA agarose was eluted with 4 volumes of elution buffer (binding buffer with a linear pH gradient of 5.0, 4.5, and 4.0). The eluate was refolded by dialysis in 500 mL of PBS containing 10% glycerol at 4℃ for 4 hr and concentrated using Amicon Ultra-4 Centrifugal Filter Unit with a molecular weight of 30 kDa (EMD Millipore, Billerica, MA, USA).
The presence of the target protein was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot. SDS-PAGE was performed according to the procedure described previously [20]. For western blotting, protein extracts separated by SDS-PAGE were transferred to a 0.45-µm pore (polyvinylidene difluoride (PVDF) membrane (Roche) by using a Semi-Dry Transfer Cell (Bio-Rad, Hercules, CA, USA). The expression of the truncated ORF2 protein was detected by immunodetection with anti-His6-peroxidase (Roche) using the manufacturer's protocol. Finally, the target protein was visualized with 3,3'-diaminobenzidine (DAB) as a substrate [21]. Concentration of the target protein was determined by the Bradford method [22]. The second truncated form of the ORF2 protein (ORF2.2) was expressed and purified using the same procedure.
4. ELISA
The diagnostic utility of the 2 purified truncated ORF2 proteins was evaluated by developing 2 ELISAs for the detection of HEV IgG antibody. Checker Board Titration and calculation of P/N value (absorbance value of the test sample/absorbance value of the negative control) were used to determine the optimal dilution of the antigen, serum, and conjugate. Briefly, ELISA plates (MAXISOR; Nunc, Roskilde, Denmark) were coated with 50 µL/well of serial dilutions of the 2 purified proteins separately (0.3-10 µg) in 50 mM carbonate/bicarbonate buffer (pH 9.6), incubated overnight at 4℃, and incubated at 37℃ for 1 hr. Two uncoated wells were used as blanks. The plates were washed 3 times with PBS containing 0.05% Tween-20 (PBST, pH 7.4) and tapped dry; then, the plates were blocked with 200 µL/well of 2% casein hydrolysate and 1% bovine serum albumin (BSA) in PBS at 37℃ for 1 hr. The well contents were emptied completely and the wash step was repeated as described above, and the wells were tapped dry. At a concentration of 100 µL/well, each serum sample was diluted in sample diluent (1% BSA in 10 mM sodium citrate buffer, pH 6.0, 0.5% Tween 20, 0.09% sodium azide), added to the wells in duplicate, and incubated at 37℃ for 1 hr. The plate was washed as described above, and 100 µL of an horseradish peroxidase (HRP)-conjugated goat anti-human IgG secondary antibody (Abcam company, Cambridge, MA, USA) diluted 1:15,000 in diluent buffer (1% BSA in 150 mM NaCl and 10 mM Tris buffer, pH 8.0, 0.05% Tween 20) was added to each well and incubated at 37℃ for 1 hr. After incubation, the plates were washed as described above. The substrate solution (100 µL/well, H2O2 and 10 mg of tetramethylbenzidine [TMB] in 1 mL of dimethylsulfoxide [DMSO] and 100 mL of citrate buffer, pH 6.0) was added, and the plates were incubated in a dark place at room temperature for 20 min. Finally, 100 µL/well of 1 M H2SO4 was added to stop the reaction.
The absorbance of the wells was read at 450 nm by an ELISA reader (Tecan Sunrise ELISA Reader; Tecan Trading AG, Mannedorf, Switzerland). The results were analyzed, and the cut-off value was determined as the mean absorbance value of 42 negative serum samples plus 2 standard deviations. All 42 negative serum samples were tested by a commercially available kit (Genlabs Diagnostics Inc., Irvine, CA, USA) for HEV IgG antibodies All 42 negative serum samples were negative for HEV IgG antibody by DIA.PRO HEV IgG ELISA kit. The 42 verified-negative serum samples were tested in duplicate in 3 separate coated plates for each in-house ELISA. On the basis of the obtained results, a cut-off value was determined for each in-house ELISA. The difference in cut-off values between the 3 plates was less than 0.02; therefore, the mean cut-off value of the 3 plates was used. After calculating the cut-off value, a pooled negative serum was prepared from the 42 negative serum samples, which was used as a negative control in the evaluation of 220 serum samples by the 2 in-house ELISA using 4 replicates each. These 220 serum samples were randomly selected from samples collected in the laboratory of Imam Khomeini Hospital in Ahvaz city. Ethical consent as written form was obtained from all participants.
The 42 negative serum samples as well as 26 weakly positive and 32 highly positive serum samples were tested to determine reproducibility (intra-assay and inter-assay precisions), sensitivity, and specificity. To determine within-run or intra-assay precision, each sample was tested in 24 replicates in a single day, and the coefficient of variation for each sample was determined. To determine between-run or inter-assay precision, each sample was tested in 8 replicates on 3 sequential days, and the coefficient of variation for each sample was determined. All serum samples were obtained from the virology reference lab of Jundishapour University. Regarding inter-assay precision, the mean coefficients of variation for the highly positive serum samples, weakly positive serum samples, and negative serum samples were 5.13%, 6.77%, and 6.18%, respectively. Regarding intra-assay precision, the mean coefficients of variation for the pooled positive serum samples and the pooled negative serum samples were 8.05% and 11.5%, respectively. Thus, there was acceptable reproducibility.
The 220 serum samples were tested for anti-HEV IgG antibodies by using the 2 in-house ELISAs and DIA.PRO HEV IgG ELISA kit. Briefly, the following steps were carried out for the 2 in-house ELISAs. All 220 serum samples and the negative and positive controls were diluted 1:21 in sample diluents, and 100 µL of each diluted sample was added in duplicate to coated wells of the ELISA plates. The ELISA plates were incubated at 37℃ for 60 min, washed 5 times with PBST, pH 7.4. One hundred microliters of diluted HRP-conjugated goat anti-human IgG secondary antibody was added to each well, and the plates were incubated at 37℃ for 60 min. The wash step was carried out as described above. One hundred microliters of chromogen-substrate mixture was added to each well, and the ELISA plates were incubated at room temperature in a dark place for 20 min. Finally, 100 µL of 1 M sulfuric acid was added to each well to stop the enzymatic reaction. The absorbance of the wells was read at 450 nm by the ELISA reader. The results of the tests were analyzed by comparison with the cut-off value. DIA.PRO HEV IgG ELISA kit for the detection of anti-HEV IgG antibodies was used according to the manufacturer's instructions.
RESULTS
1. Optimization of expression of the truncated ORF2 gene
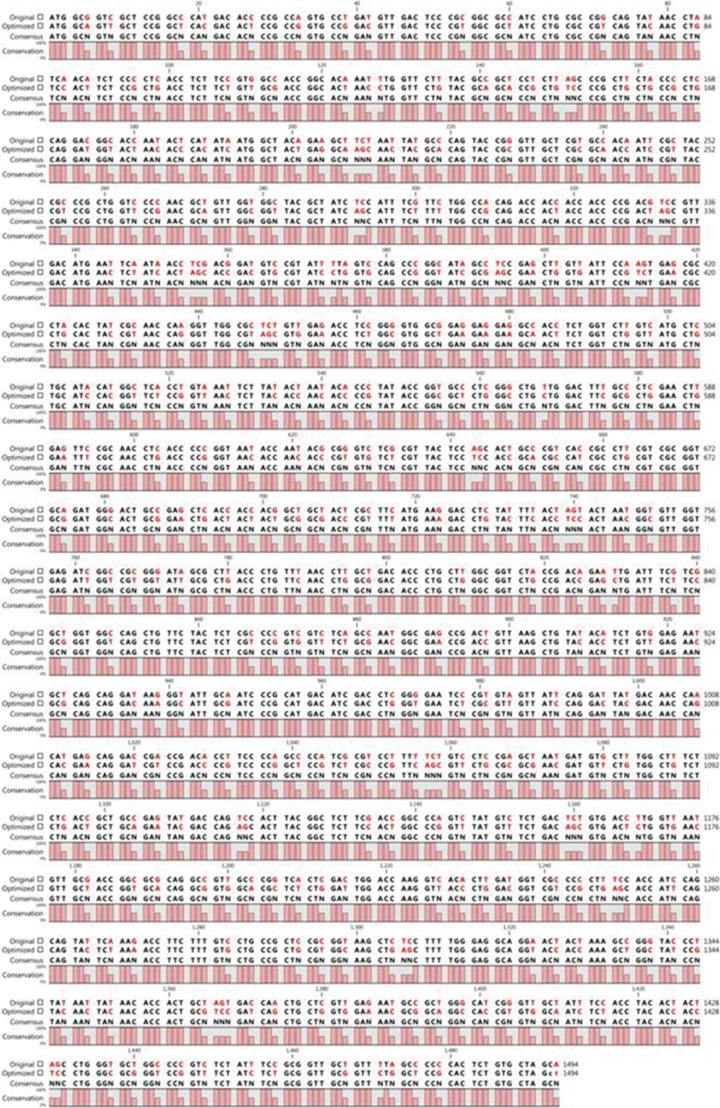
The truncated ORF2 gene was optimized for expression in E. coli by using GenScript software (GenScript USA Inc.) without changing the amino acid sequence by analyzing the codon adaptation index (CAI) before and after optimization. The level of protein expression is correlated with the CAI value, where a CAI>0.8 is ideal for expression in the target host. In our study, a CAI value of 0.85 was determined for the optimized gene. The sequences of the native ORF2 gene and the optimized gene are shown in Fig. 1.
2. Construction of the recombinant plasmid
After subcloning, the recombinant plasmids were confirmed by PCR, restriction enzyme digestion, and DNA sequencing. PCR amplification using universal plasmid primers followed by electrophoresis on 1.2% agarose gel revealed a PCR product of 1,943 bp for ORF2.1 and 1,750 bp for ORF2.2 (Fig. 2).
3. Protein expression and purification
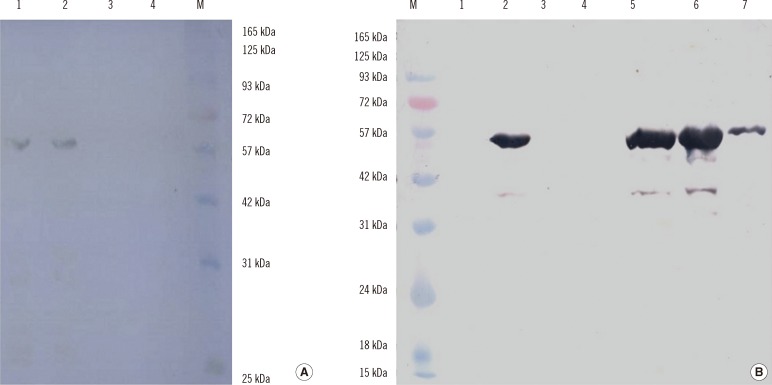
The optimal conditions for protein expression were induction with 1 mM IPTG with shaking at 37℃ for 4 hr. The highly expressed target proteins were purified by Ni2+-chelate-affinity chromatography (Qiagen). The concentration and purity of the proteins were maximized by Amicon Ultra-4 Centrifugal Filter Unit (EMD Millipore) with a molecular weight of 30 kDa. The expression and purification of the truncated ORF2 protein was confirmed by SDS-PAGE and western blotting, which revealed a protein band of approximately 60 kDa for ORF2.1 and approximately 55 kDa for ORF2.2 (Fig. 3). The abundant target proteins were eluted with the elution buffer at pH 4.5. The yield of the purified protein was approximately 1 mg/L of the culture medium. Western blotting was carried out to confirm the presence of the ORF2.1 and ORF2.2 proteins in E. coli BL21 (DE3) (Fig. 4).
4. ELISA based on the 2 truncated ORF2 proteins
The ELISA was optimized by examining different antigen concentrations and serum dilutions. The optimal concentration of the coating ORF2.1 protein for detecting HEV IgG antibody was 2 µg/mL. The optimal blocking buffer was 2% casein and 1% BSA in PBS. The optimal serum dilution was 1:21, and the optimal conjugate dilution was 1:15,000. The optimal conditions for the ORF2.2 protein were identical. The cut-off values for ORF2.1 and ORF2.2 were 0.35 and 0.4, respectively. Samples were considered positive, if the OD was above the cut-off value. The 220 serum samples were tested anti-HEV IgG antibodies by using the 2 in-house ELISAs and the results were compared with DIA.PRO HEV IgG ELISA kit (Fig. 5). Among the 220 serum samples analyzed by the 2 in-house ELISAs, 21 were positive for anti-HEV IgG antibodies; 23 of the samples tested positive by using DIA.PRO HEV IgG ELISA kit (Table 1). The 2 in-house ELISAs had 98.1% concordance, 99.5% negative agreement, and 87% positive agreement compared with DIA.PRO HEV IgG ELISA kit (kappa=0.899 and P=0.625). For both in-house ELISAs, the same 21 samples tested positive, while the remainder of the samples tested negative. Thus, there was 100% concordance in the detection of anti-HEV IgG between the in-house ELISA coated with ORF2.1 protein and that coated with ORF2.2 protein (kappa=1.00 and P=1.00).
There were 4 serum samples with different results between the 2 in-house ELISAs and DIA.PRO HEV IgG ELISA kit. For 3 of the 4 samples, the OD values were close to the cut-off value for the in-house ELISA based on ORF2.1. For the other in-house ELISA, the OD values of these 3 samples were below the cut-off value. Therefore, these 3 samples were considered negative in both in-house ELISAs. The OD of those 3 samples was considered weakly positive by DIA.PRO HEV IgG ELISA kit. One sample was determined to be negative by DIA.PRO HEV IgG ELISA kit, but was found to be positive by the 2 in-house ELISAs; this sample was highly lipemic.
DISCUSSION
HEV antibodies are indicative of past infection. For seroepidemiological studies, recombinant ORF2 protein is needed in order to detect HEV antibodies. Many studies have investigated low-cost methods for over-expressing recombinant proteins as diagnostic tools [10, 11, 23]. In our study, 2 truncated forms of HEV ORF2 protein were produced on a large scale for the development of 2 ELISAs that could detect anti-HEV IgG. The truncated forms (especially the 56-kDa form) of ORF2 protein are more stable and useful for the detection of antibodies against HEV [11, 15, 16]. Therefore, we expressed and purified 2 truncated forms of the ORF2 protein in E. coli.
The advantages of expressing proteins in E. coli are cost effectiveness, ease of handling, short reproductive cycle, and low pollution [24]. However, expressing proteins in E. coli can result in low-level or failed expression because of some problems including high GC content and rare codon in the gene sequence [24]. To overcome poor expression, we optimized our gene sequence for expression in E. coli; this provided several advantages, including elimination of polyadenylation sites, splicing sites, killer motifs, repeated sequences, RNA secondary structure, and high GC content [25, 26]. The expression level can be improved through using host-favorite codons and lowering the GC content, since rare codons and high GC content can decrease or even prevent expression [27]. There are several approaches for expressing the HEV structural gene [21, 28]. However, no previous study has utilized codon-optimized expression of the ORF2 protein. Our results indicate that codon optimization resulted in effective expression in E. coli with high protein yield.
For the positive serum samples, the mean OD values were higher when tested by the in-house ELISA coated with ORF2.2 as compared to the in-house ELISA coated with ORF2.1 and DIA.PRO HEV IgG ELISA kit. When tested by the in-house ELISA coated with ORF2.1 protein, the mean OD values of the negative serum samples were close to the cut-off value for this ELISA. This is a limitation of this in-house ELISA as compared with the in-house ELISA coated with ORF2.2 protein and DIA.PRO HEV IgG ELISA kit.
Since E. coli was used to express these 2 proteins, the proteins could have been contaminated with bacterial proteins during purification. Subsequently, this contamination could have led to false-positive results for serum samples containing antibodies against E. coli. To eliminate this possibility, Toxin Eraser Endotoxin Removal kit (Genscript Inc.) was used to remove bacterial proteins from the expressed proteins.
An approximately 4 g of bacterial pellet was obtained from each liter of culture media, which yielded approximately 1 mg of purified protein. One hundred nanograms of the purified protein per well was used in each in-house ELISA to detect anti-HEV IgG. It is estimated that approximately 10,000 serum samples could be screened using the protein obtained from 1 L of bacterial culture. In conclusion, high yields of the highly purified proteins were obtained and could be used as a diagnostic antigen in ELISA-based detection of anti-HEV IgG in serum. Moreover, both in-house ELISAs are highly concordant with DIA.PRO HEV IgG ELISA kit.




 XML Download
XML Download