

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The incidence of venous thromboembolism (VTE) is high among Caucasians, and some individuals and families are predisposed to developing it. Thrombophilia predisposes an individual to developing thrombosis [1, 2]. In Western countries, a large number of VTE patients have been found to be carriers of a coagulation factor V polymorphism, factor V Leiden (R506Q) [3-8]. Factor V Leiden (R506Q) has normal coagulation activity but shows resistance to the activated protein C (APC)-anticoagulant system (APC resistance). Thus, it can cause excessive blood coagulation. Another influential factor that has a different mechanism of action has also been discovered: a single-base substitution in the 3'-untranslational region of the prothrombin gene, prothrombin G20210A. Carriers of this mutation are prone to developing VTE [9]. The majority of Caucasian VTE patients have 1 of these 2 thrombophilias: factor V Leiden (R506Q) and prothrombin G20210A [7-11].
Meanwhile, carriers of these polymorphisms are quite rare among non-Caucasians [12-18], thus warranting the assumption that very few non-Caucasians are affected by VTE. However, recent studies demonstrate that there are a considerable number of VTE patients among non-Caucasians such as the Japanese and Chinese [13-21].
1. Thrombophilia and VTE in Japan and other Asian countries
After the discovery of factor V Leiden (R506Q) [3-5], Shen et al. from National Taiwan University reported that 47 (55%) out of 85 thrombosis patients had reduced activity of the APC anticoagulant system (28 had low protein S activity, 16 had low protein C activity, and 3 had both low protein S and C activities) [15]. Our investigation on Japanese subjects revealed that 49 (58%) of 85 deep vein thrombosis (DVT) patients had reduced activity of factors of the APC anticoagulant system (22 had low protein S activity, 9 had low protein C activity, and 18 had both low protein S and C activities) and that the reduced activities of 27 patients were due to genetic abnormalities in the protein S and C molecules [18]. However, no carriers of factor V Leiden (R506Q) were found among these Japanese patients [18]. A report from Hong Kong claims that as many as 53% of Chinese VTE patients have reduced activity of the APC anticoagulant system [19]. All of these studies provide very similar results, suggesting that Japanese and Chinese individuals have thrombophilias that differ from those of Caucasians, with a high likelihood of thrombophilia being a protein S or C molecule abnormality-especially protein S molecule abnormality, at least in Japan. These results are summarized in Table 1.
VTE gained much public attention and acquired the layman's term of "economy class syndrome" recently [22-24]. This kind of VTE develops because of the stagnation of blood flow in the deep veins of a lower limb as a result of prolonged sitting in the same position in a confined space (i.e., an economy-class seat) of an aircraft. This condition, diagnosed as DVT and pulmonary embolism, has been reported to occur in people forced to live in emergency shelters as a result of natural disasters such as the earthquake and tsunami that occurred near the northeastern coast of Japan on March 11, 2011 [25].
2. Protein S Tokushima (K155E) in Japanese DVT patients and healthy individuals
Protein S Tokushima (K155E), an abnormal protein S molecule, was discovered in thrombotic patients by Shigekiyo et al. [26] and Yamazaki et al. [27]. The lysine (K) residue at position 155 of the protein S Tokushima molecule is replaced by glutamic acid (E) [27, 28]; the molecule has low protein S activity [26, 28]. We previously identified a patient suffering from DVT as a homozygous carrier of protein S Tokushima (K155E) [18]. The protein S activity and free protein S concentration in this patient were 35% and 78% of the reference value, respectively; thus, the specific activity (activity/protein concentration) of protein S Tokushima was 45% [18]. The specific activity of protein S Tokushima (K155E) expressed in HEK293 cells was slightly less than 60% of the specific activity of the wild type [29]. The extent of reduction between the in vivo activity and activity in the cultured cell expression system differed slightly. Nevertheless, the activity of protein S Tokushima (K155E) was obviously reduced.
The frequency of heterozygous carriers of protein S Tokushima (K155E) among healthy individuals in Japan is nearly 2% [18, 21, 30, 31], namely, 77 heterozygous carriers among 4,319 individuals [30], indicating a mutant allele frequency of 0.0089. The frequency is much higher (about 6-10%) among DVT patients, with an odds ratio of 3.74-8.56 [18, 21, 30, 31]. Whether protein S Tokushima (K155E) occurs in other Asian countries is an important aspect of mapping thrombophilia among Asians, and international surveys are needed to determine this.
3. Variants in the protein S gene (PROS1) in Japanese DVT patients
In our previous studies [18, 32], the age at first incidence of DVT was unexpectedly low, peaking from 20-30 yr for both men and women. Surprisingly, the first incidence occurred before the age of 40 yr in about 60% of the patients. This suggests that an individual's constitutional factors influence the development of DVT more strongly than lifestyle factors.
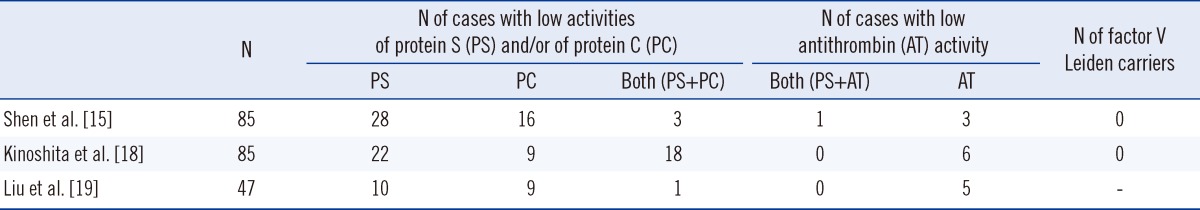
Combined with our analysis [18, 33-36] and the results of other studies [37-48], a total of 51 variants were identified in a Japanese population. These variants are distributed throughout the coding sequence (exons 2-15), except exon 1, of the protein S gene, PROS1 (Fig. 1) [49]. The close sequence homology between PROS1 and its pseudo-gene, PROS2, suggests the possibility of gene rearrangements similar to those in the von Willebrand factor (vWF) gene [50, 51]. However, to our knowledge, no recombination between these 2 genes has been described. A large deletion of PROS1 has been found in Swedish families in which mutations in PROS1 were not detected despite sequencing [52, 53], suggesting that screening for large deletions in PROS1 may be useful for protein S deficiency patients.
 | Fig. 1Structural model of protein S and its variants (Courtesy of Dr. Yoshito Abe, Laboratory of Protein Structure, Function and Design, Graduate School of Pharmaceutical Sciences, Kyushu University). Variants of the protein S molecule observed in our laboratory are shown by space-filling symbols in individual domains of the Gla domain (A), EGF domain (B), and SHB domain (C). Since only the EGF3-4 domain structure of protein S was determined [58], the other domains were prepared by homology modeling using the Swiss-model web server [59]. The coordinates of the structures of the Gla domain of factor IX [60], EGF domain of blood coagulation factor VIIA [61], and laminin G-like domain of Gas6 [62] were used as templates for homology modeling. The whole structural model of protein S was constructed by connecting each domain corresponding to an amino acid sequence.
|
4. Role of the APC anticoagulant system in coagulation control in vivo
The anticoagulant system in a healthy body mainly comprises 3 systems: 1) the tissue factor pathway inhibitor (TFPI) anticoagulant system, 2) antithrombin (AT) anticoagulant system, and 3) APC anticoagulant system (Fig. 2). Both the TFPI and AT anticoagulant systems have very potent anticoagulant activity. It is well known that the TFPI anticoagulant system plays its physiological and pathological roles through the inhibition of tissue factor-initiated blood coagulation, Meanwhile, the AT anticoagulant system plays these roles through the inhibition of thrombin and coagulation factor Xa. Unlike the other 2 anticoagulant systems, the APC anticoagulant system is only activated after thrombin is formed as a result of the activation of the coagulation system. The APC anticoagulant system is unique in that its anticoagulation activity is regulated in proportion to the activity of the coagulation system (Fig. 2). Thus, the APC anticoagulant system regulates the balance between coagulation and anticoagulation activities. Abnormal thrombus formation is thought to occur when the equilibrium between the coagulation system and APC anticoagulant system is disturbed [49, 54].
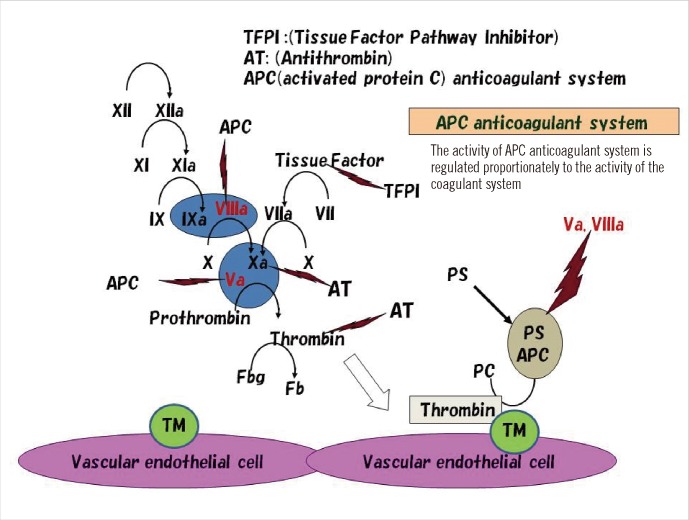 | Fig. 2Coagulation and anticoagulation systems. The anticoagulant systems of TFPI and AT directly inhibit tissue factor-initiated blood coagulation and coagulation factor Xa/thrombin, respectively. In the APC anticoagulant system, when thrombin is produced by the coagulation system, it forms a complex with thrombomodulin (TM) on the surface of the vascular endothelium and loses its activity to convert fibrinogen (Fbg) to fibrin (Fb) and instead converts protein C to APC. With the help of protein S (PS), the APC/PS complex inhibits coagulation factors Va and VIIIa. Thus, the activity of the APC anticoagulation system is regulated in proportion to the activity of the coagulation system.
|
Summarizing the results from surveys and existing research allow us to conclude the following. Thrombophilia among Caucasians is mainly caused by resistance to the APC anticoagulant system (APC resistance) and factor V Leiden (R506Q) [3-8, 10, 11], while thrombophilia among Japanese and Chinese individuals is due to the reduced activity of the APC anticoagulant system (APC dysfunction) [15, 17-21, 31, 49, 54].
These 2 phenomena are not in fact all that unpredictable: either the coagulation activity becomes relatively stronger than the APC anticoagulant activity due to factor V Leiden (R506Q) or the APC anticoagulant activity declines relative to the coagulation activity due to molecular abnormalities in protein S or C. Taken together, these findings suggest that the APC anticoagulant system maintains a balance between coagulation and anticoagulation activities, thus greatly contributing to thrombus formation. Regardless of APC resistance in Caucasians or APC dysfunction in Japanese and Chinese individuals, "the creation of a condition where coagulation activity becomes relatively stronger than the APC anticoagulant activity" could be the trigger mechanism for thrombosis development in thrombophilic carriers [49, 54].
5. Significance of the measurement of protein S specific activity and its practical use
Protein S deficiency is approximately 10 times more prevalent in Asians than in Caucasians [18]. In addition, the prevalence of the type II deficiency is quite high, at least in Japan [18, 21, 31, 49]. To screen for type II protein S deficiency, clotting-based protein S activity assays and free protein S assays are currently performed. However, Kimura et al. report that these assays are unsuitable for identifying deficiencies such as protein S Tokushima (K155E) [55]. A new quantitative protein S assay method with the following advantages was recently developed [56]: 1) total protein S, i.e., the sum of free protein S and bound protein S, can be measured; 2) the accuracy and reproducibility of the measurement is dramatically improved because protein S can be measured without separating the free form from the bound form; 3) the absolute amount (µg/mL) of protein S can be determined; and 4) the specific activity of the protein S molecule can be calculated by measuring the protein S activity and amount of protein S. The type II deficiency can easily be determined by measuring the specific activity of the molecule [56].
The protein S activity and amount of protein S (mean±2SD) in men (N=107) were 25.7±6.8 µg/mL protein S equivalent and 26.0±6.8 µg/mL protein S, respectively, while those in women (N=94) were 21.9±6.8 µg/mL protein S equivalent and 22.4±6.4 µg/mL protein S, respectively, confirming the difference in protein S between sexes. However, the mean protein S specific activities and its reference intervals (mean, mean±2SD) were 0.99 and 0.79-1.19 in men (N=107), respectively, and 0.98 and 0.76-1.20 in women (N=94), respectively, showing no difference between the sexes [56]. These results indicate that estrogen, which is secreted more in women than in men, suppresses protein S production [57]; but, the protein S molecules produced in both sexes are normal, and thus there is no difference in the protein S specific activity between sexes. This quantitative protein S assay can rapidly identify carriers of protein S type II deficiency without genetic testing by measuring the total amount of protein S, total protein S activity, and protein S specific activity in the blood.
Go to : 
CONCLUSION
Thrombophilias among Japanese and Chinese individuals are mainly due to APC dysfunction, whereas their major cause in Caucasians is APC resistance [49, 54]. Whether APC dysfunction occurs in other Asian countries is an important unresolved aspect of thrombophilia among Asians; international surveys are needed to determine this. A newly developed assay system for the specific activity of protein S would be useful for such international surveys, which could potentially contribute to the early detection of thrombophilic traits.
Go to :
 XML Download
XML Download