

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mucopolysaccharidosis III (MPS III), also known as Sanfilippo syndrome, is an autosomal recessive disorder with symptoms that reflect the inability to degrade heparan sulphate. The 4 subtypes of the disease are defined by enzyme deficiency: MPS IIIA (heparan N-sulfatase; EC 3.10.1.1; Mendelian Inheritance in Man [MIM] # 252900), MPS IIIB (α-N-acetylglucosaminidase; EC 3.2.1.50; MIM# 252920), MPS IIIC (heparan acetyl-CoA: alpha-glucosaminide N-acetyltransferase [HGSNAT]; EC 2.3.1.78; MIM# 252930), and MPS IIID (N-acetylglucosamine 6-sulfatase; EC 3.6.14; MIM# 252940) [1]. Prevalence studies estimate that MPS IIIC occurs in 0.21 per 100,000 live births in The Netherlands, 0.17 in Sweden, 0.12 in northern Portugal, 0.07 in Australia, and 0.03 in Taiwan [2-6].
Although phenotypic variation has been observed, the symptoms of MPS IIIC are similar in all subtypes. Most MPS IIIC patients have severe clinical manifestations with onset in infancy or early childhood. They rapidly develop progressive and severe neurological deterioration that causes hyperactivity and sleep disorders accompanied by behavioral abnormalities, neuropsychiatric problems, mental retardation, hearing loss, and visceral manifestations such as mild hepatomegaly, mild dysostosis multiplex, mild coarse face, and hypertrichosis [7]. In the very rare MPS IIIC patients who experience symptom onset during adulthood, disease progression is similar to the forms with childhood onset with regard to severity and time course [8]. The initial diagnosis of all types of MPS III is based on increased concentrations of heparan sulfate in the urine. Enzymatic assays of leukocytes and/or fibroblasts confirm the diagnosis and permit discrimination among the different disease subtypes.
The gene encoding HGSNAT is located on chromosome 8p11.1 and contains 18 exons. The cDNA encodes a product of 635 amino acids, of which the N-terminal 30 amino acids are predicted to form a cleavable signal peptide. Along the remainder of the protein, there are 11 transmembrane domains, and up to 5 N-linked glycosylation sites [9, 10]. More than 54 mutations have been identified in the HGSNAT gene (Human Gene Mutation Database, www.hgmd.org), but there have been no reports of MPS IIIC in Korea. Although previous reports have described Korean patients with MPS, most of them presented with MPS type II [11-15]. MPS III accounts for approximately 18% of Korean patients with MPS, and most of them have MPS IIIA and MPS IIIB [16]. In the present study, we present biochemical and genetic analyses of a Korean patient presenting with the MPS IIIC phenotype, who was screened for mutations in the HGSNAT gene.
Go to : 
CASE REPORT
1. Clinical findings
A female patient was referred to our hospital at the age of 2, after presenting with hepatomegaly and delayed motor development. The parents were healthy, non-consanguineous ethnic Koreans. She was the second child in her family and had an older brother exhibiting a normal phenotype. Her height was 88.2 cm (10-25th percentile), and she weighed 13.1 kg (25-50th percentile). On physical examination, the patient appeared to have mildly coarse facial features and a distended abdomen. The nasal bridge was low and the eyebrows were prominent, broad, and straight. Her cognitive function and speech development were normal. Lateral view of the spine showed vertebral dysplasia with ovoid-shaped vertebral bodies, without the evidence of vertebral breaking (Fig. 1A). Radiographs of both hands revealed widening of the metacarpals and the proximal ends of the phalanges (Fig. 1B). Separation between the 3rd and 4th finger was observed in the left hand, and these findings were suggestive of mucopolysaccharidosis. Serum levels of alanine transaminase, aspartate aminotransferase, calcium phosphate, alkaline phosphatase, and creatinine were normal. The amino acid profile and organic acid profile did not show any abnormalities.
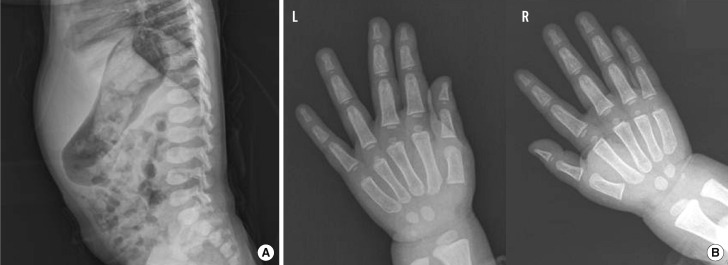
2. Biochemical analyses
We measured urinary glycosaminoglycan (GAG) levels using the cerylpyridinium chloride (CPC) precipitation test. CPC reagent was added to the centrifuged random urine sample and the standard (chondroitin sulfate, 100 mg/L). The absorbance at 680 nm was read after 10 min and compared with that of the standard. The GAG/creatinine ratio was used to measure the urinary excretion of GAG. The urinary GAG level of the patient was 984.4 mg GAG/g creatinine, which is elevated when compared with normal reference levels (reference range: <175 mg GAG/g creatinine). Additionally, we performed thin-layer chromatography for the urine. The elevated GAG was identified as heparan sulfate by thin-layer chromatography, which suggested MPS III. To confirm the MPS diagnosis and to determine the disease subtype, MPS III enzymatic assays including the evaluation of HGSNAT activity were performed as described by Voznyi et al. using the artificial substrate 4-methylumbelliferyl β-D-glucosaminide (Moscerdam, Rotterdam, The Netherlands) [17]. The enzyme activity was measured in sonicated fresh leukocytes and compared with that of the normal controls. The HGSNAT activity of the patient was 0.7 nmol/17 hr/mg protein (reference range, 8.6-32 nmol/17 hr/mg protein), and there was no decrease in the enzyme activity for MPS IIIA, IIIB, and IIID (Table 1).

3. Molecular genetic analysis
Blood samples were collected from the patient after informed consent was obtained from the parents. Genomic DNA was isolated from peripheral blood leukocytes using a Wizard genomic DNA purification kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. The 18 exons of the HGSNAT gene, along with their flanking intronic regions, were amplified by PCR by using primers designed by the authors (sequences available upon request) and a thermal cycler (Model 9700; Applied Biosystems, Foster City, CA, USA). The amplification product (5 µL) was treated with 10 U shrimp alkaline phosphatase and 2 U exonuclease I (USB Corp., Cleveland, OH, USA). Direct sequencing of the DNA was performed using the ABI Prism 3100 Genetic Analyzer (Applied Biosystems) with the BigDye Terminator Cycle Sequencing-Ready Reaction Kit (Applied Biosystems). The cDNA nucleotide sequences of the gene examined were numbered according to their respective GenBank accession numbers of NM_152419.2 for HGSNAT. The mutation nomenclature used here follows the recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/), with nucleotide +1 corresponding to the A of the ATG translation initiation codon.
The patient was found to have compound heterozygous mutations for c.234+1G>A and c.1150C>T of HGSNAT. The patient's mother was heterozygous for the c.234+1G>A mutation and her father was heterozygous for the c.1150C>T mutation. The c.234+1G>A mutation leads to an early termination after a frame shift by changing G to A at c.234+1 of intron 2, thereby prompting an amino acid substitution from Asp to Val (p.D40Vfs*19). The c.1150C>T mutation is a change from C to T at the 1150th base of the 12th exon to T and leads to the substitution of arginine with a premature termination codon (p.R384*). These nonsense and splice site mutations have both been reported previously [18] (Fig. 2).
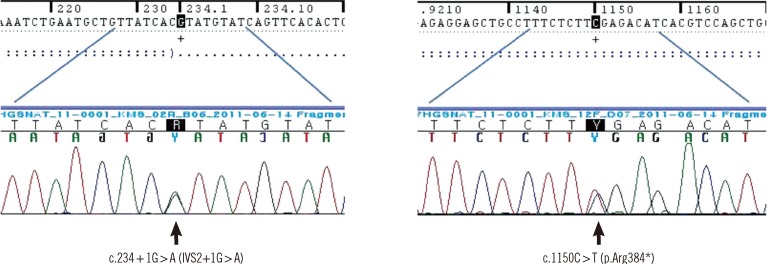
Go to :
DISCUSSION
MPS IIIC is a rare disease, and its genetic background was identified only 5 yr ago [9, 10]. Thus, only a few studies on MPS IIIC mutations have been published [9, 10, 18, 19]. To the best of our knowledge, this is the first report of genetically confirmed MPS IIIC in Asia. In agreement with previously published data, the patient in this study showed classic signs or symptoms of MPS IIIC, such as coarse facial features, dysostosis, and hepatomegaly, although information on the natural course of MPS IIIC is limited [7, 18, 20].
In the mutation study, our patient was found to be a compound heterozygote for 2 known mutations. The c.234+1G>A mutation, located in intron 2, was first described in 1 Spanish and 2 Moroccan patients [10, 20]. This mutation has been shown to cause exon skipping, thereby resulting in the deletion of a substantial portion of the protein [10]. The most common splice site mutation, c.234+1G>A, was identified in 7 patient families: 1 from France, 1 from Italy, 1 from Spain, 1 from Turkey, 2 from Morocco, and 1 from Canada (in which both parents are of Moroccan origin) [10, 18-21].
The c.1150C>T (p.R384*) nonsense mutation has been found to occur both homo- and heterozygously. All detected mutations have significant structural and functional impacts on the enzyme and RNA [18]. A recent study showed that transcripts carrying premature termination codons are rapidly degraded via the nonsense-mediated mRNA decay pathway, which protects the cell from potentially harmful effects of truncated proteins [22]. The p.R384* mutation was detected in 10 patients from 6 different countries (Poland, Czech Republic, Italy, The Netherlands, Canada, and Turkey) [10, 18, 19, 21]. Feldhammer et al. showed that these mutations had relatively high frequencies among MPS IIIC families and were also characterized by wide distribution, which did not suggest a founder effect in any particular population [19]. The present case supports the previous studies showing that these mutations appear to be common in different populations. Our patient had severely reduced heparan acetyl-CoA: α-glucosaminide N-acetyltransferase activity that was thought to be caused by 2 HGSNAT mutations.
The natural course of MPS III consists of devastating and irreversible neurodegenerative symptoms, including mental retardation and behavioral problems. Therefore, the main focus of treatment is the central nervous system. Although no effective therapy is yet available for MPS III, several promising therapeutic approaches are under investigation [7]. For the successful application of appropriate therapies, the subtype of MPS III should be clearly defined. Because the clinical signs, symptoms and course of the disease are indistinguishable among MPS III subtypes, a confirmatory diagnosis should be made on the basis of biochemical and genetic analyses.
In summary, we identified MPS IIIC in a Korean patient by using clinical, biochemical, and molecular analyses. Such HGSNAT mutation studies are valuable for genetic confirmation, family study, and carrier testing. Further studies are required to understand the molecular spectrum in Korean patients afflicted with MPS IIIC.
Go to :
 XML Download
XML Download