

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Parents who are carriers of balanced translocations are at risk of producing gametes with unbalanced translocations. The type of unbalanced translocation is dependent upon the mode of segregation. A 2:2 segregation event (adjacent-1 or adjacent-2) would result in gametes with a modal number of 46, but could yield partial trisomy/monosomy of the chromosomes involved in the translocation. In contrast, a 3:1 segregation usually occurs when one of the derivative chromosomes is very small. Displacement of the small derivative chromosome results in gametes with modal numbers of 45 or 47. These meiotic products may or may not be observed depending on the viability of the resulting offspring [1].
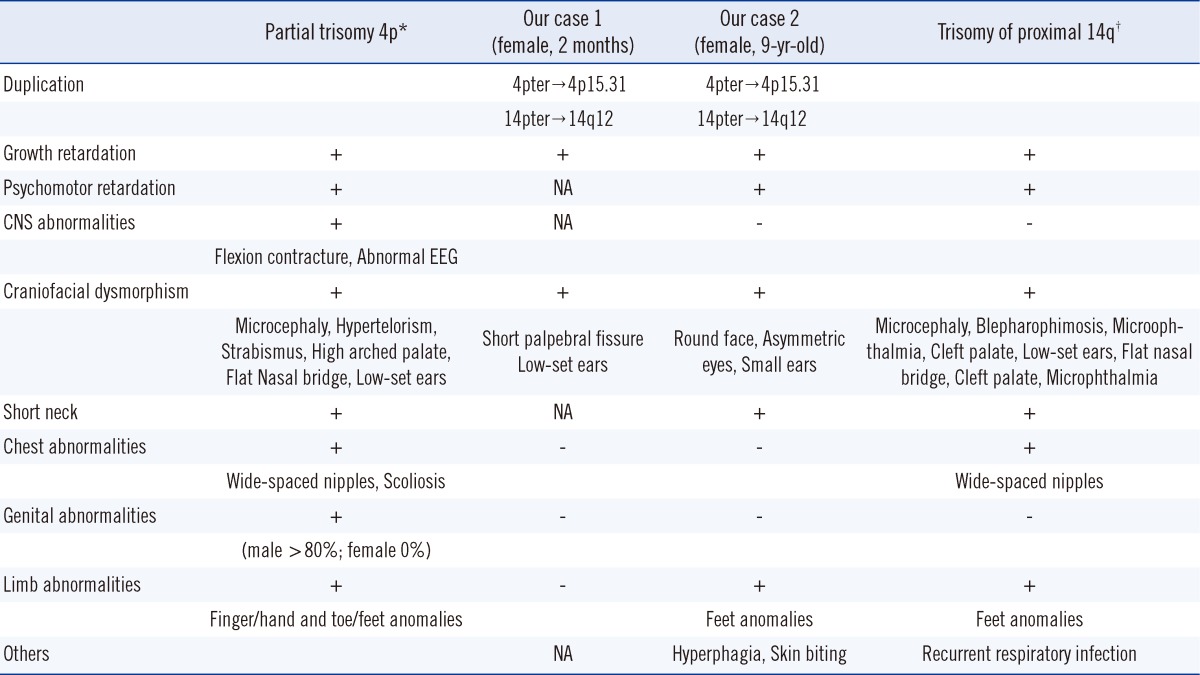
Here, we present the cases of 2 patients with partial trisomy 4p and partial trisomy 14q resulting from a 3:1 segregation of a maternal balanced t(4;14) translocation. Both patients had an extra derivative chromosome 14 made up of 4p and proximal 14q segments. Both patients showed prominent growth retardation in addition to psychomotor developmental delay. A few minor phenotypic abnormalities, including short palpebral fissure, low-set ears, a round face, asymmetric eyes, small ears, a short neck, and finger/toe abnormalities were also observed in these patients.
CASE REPORT
1. Case 1
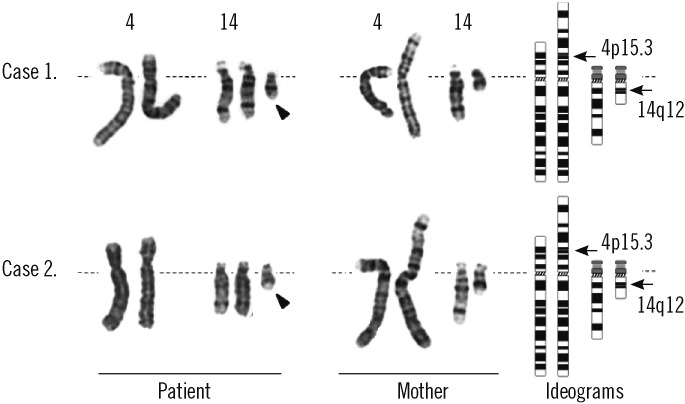
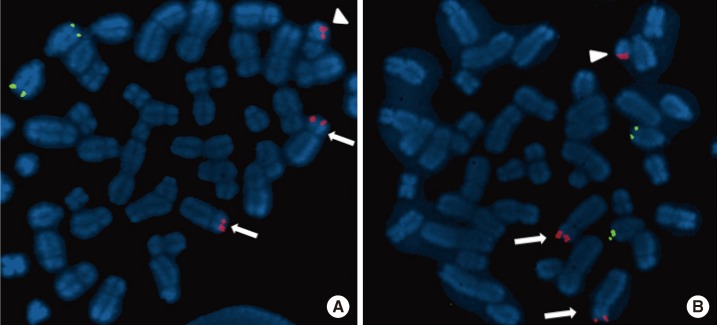
The patient was a 2-month-old girl who was admitted due to persistent diarrhea for 3 days. She was born to a 31-yr-old father and a 30-yr-old mother via vaginal delivery at 38 gestational weeks and 3 days. During pregnancy, no specific problems were identified. At birth, her weight was 2.6 kg and no specific findings were detected. She was her parent's first child, and her mother's gestational history was normal (G1-P1-A0). At the time of admission, her weight was 3.3 kg (<3rd percentile), her height 48 cm (<3rd percentile), and her head circumference 34.5 cm (<3rd percentile). The only facial abnormalities observed were a relatively short palpebral fissure and low-set ears. Her fingers and toes were long and thin, but these findings were not notable. A brain magnetic resonance imaging (MRI) revealed no abnormalities. Chromosomal analysis revealed a 47,XX,+mar[44]/46, XX[6] karyotype (GTG banding, 550 bands) (Fig. 1). In order to identify the marker chromosome, a family study was performed, and this revealed the same chromosomal complement t(4;14) (p15.31;q12) in her mother (Fig. 1). An additional array analysis using Affymetrix Cytogenetics 2.7M array (Affymetrix, Santa Clara, CA, USA), detected an abnormal approximately 19 megabase (Mb) addition from 4pter to p15.31 (19,350,350 bp point) and an approximately 31 Mb addition from 14pter to q12 (31,088, 843 bp point) (Fig. 2). To validate these results, we performed FISH using a probe for 4p16.3 (Abbott Laboratories, Abbott Park, IL, USA), which revealed an additional 4p signal on the marker chromosome (Fig. 3).
2. Case 2
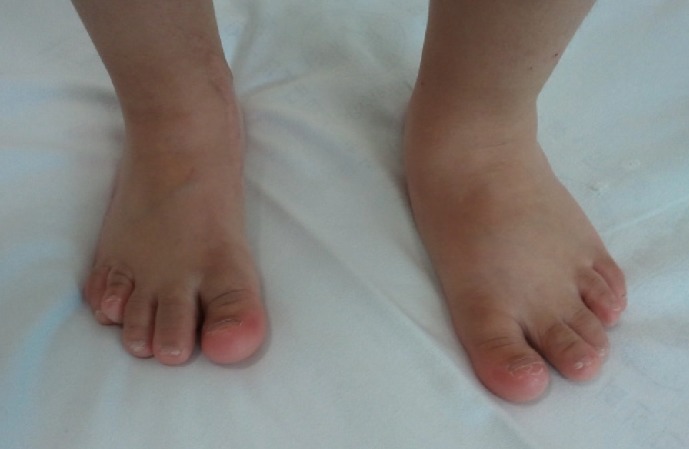
The patient was a 9-yr-old girl who was admitted for growth retardation. She was born at 39 gestational weeks and 5 days at a local hospital with no specific problems identified during the pregnancy. At birth, her body weight was 2.6 kg, but other detailed birth information could not be acquired. At age 6, she visited our hospital for growth retardation. Her weight was 16.5 kg (3rd-10th percentile) and her height was 95 cm (<3rd percentile). At that time, her parents told that she had Down syndrome based on a previous genetic study. When the patient was re-admitted to our hospital at 9 yr of age, she had a body weight of 20.8 kg (3rd-10th percentile), a height of 101.7 cm (<3rd percentile), and a head circumference of 50.2 cm (<3rd percentile). She also presented with developmental delays that included a delayed first step (at age 5 yr), mental retardation, and behavioral problems, such as skin biting and overeating. She had a round face, asymmetric eyes (smaller left eye), small ears, and a short neck. The 4th toes on both sides were short, and both big toes were relatively thick (Fig. 4). No other visible abnormalities were detected. A chromosomal study was performed to reevaluate the diagnosis of Down syndrome. In the chromosome study, 47,XX,+mar was detected in all metaphase cells from the patient, and from a subsequent family study (GTG banding, 550 bands), her mother was identified as a balanced translocation carrier with t(4;14)(p15.3;q12) (Fig. 1). Affymetrix cytogenetic array analysis detected an approximately 19 Mb addition from 4pter to p15.31 (19,351,869 bp point) and an approximately 31 Mb addition from 14pter to q12 (31,098,692 bp point) in the patient (Fig. 2). FISH analysis, using a probe for 4p16.3, revealed an additional 4p signal on the marker chromosome (Fig. 3). The patient was started on hormonal therapy, and after 4 months of therapy, her weight was recorded as 22.3 kg (3rd-10th percentile) and her height was 105.4 cm (<3rd percentile).
DISCUSSION
Trisomy 4p syndrome was first reported as a distinct clinical entity approximately 40 yr ago [2]. This syndrome is characterized by severe mental retardation, post and/or prenatal growth retardation, and mild facial dysmorphism [3]. Partial trisomy 4p can occur as a result of unbalanced meiotic segregation from a parental balanced translocation, a pericentric inversion, or a de novo duplication [3-6]. Majority of these cases are due to a parental reciprocal balanced translocation and are consequently accompanied by monosomy of the partner chromosome. The variable size of the duplicated 4p segment and the accompanying monosomy of the partner chromosome result in variable clinical findings and make the clinical diagnosis of trisomy 4p syndrome difficult.
Rarely, trisomy 4p cases appear as an additional marker chromosome [7, 8]. This additional chromosome is a derivative chromosome resulting from a 3:1 segregation of a parental balanced translocation, and consequently accompanies partial trisomy of a partner chromosome. Dallapiccola et al. previously reviewed cases of trisomy 4 and reported that 6 of 32 cases had a modal number of 47 and that most of these cases had translocations of an acrocentric chromosome, particularly chromosome 22 [9]. Another case by Takeno et al. also exhibited a rearrangement of acrocentric chromosome 13, resulting in an extra der(13)t(4;13) [6].
Partial trisomy 14 was also described in several reports from 40 yr ago [10-12]. Most of these patients were offspring of carriers of balanced translocations. The characteristic phenotypes associated with proximal trisomy 14 are developmental and growth retardation, minor facial anomalies, microcephaly, and failure to thrive. Like partial trisomy 4p, partial trisomy 14q does not show specific phenotypic characteristics indicative of distinct 14q trisomy.
Both of our cases had extra der(14) chromosomes consisting of proximal 4p (pter to p15.31) and proximal 14q (pter to q12). This der(14) is small, and similar to chromosome 21 in morphology and size (Fig. 2). This similarity resulted in an inaccurate diagnosis of Down syndrome for the patient described in case 2, and this was not questioned prior to re-examination at our hospital. When a derivative chromosome with a satellite is small, visual identification of its origin can be difficult, and it can easily be mistaken for one of the G group chromosomes (21 or 22) as in this case. Therefore, careful consideration is needed when examiners observe a new, small, acrocentric chromosome.
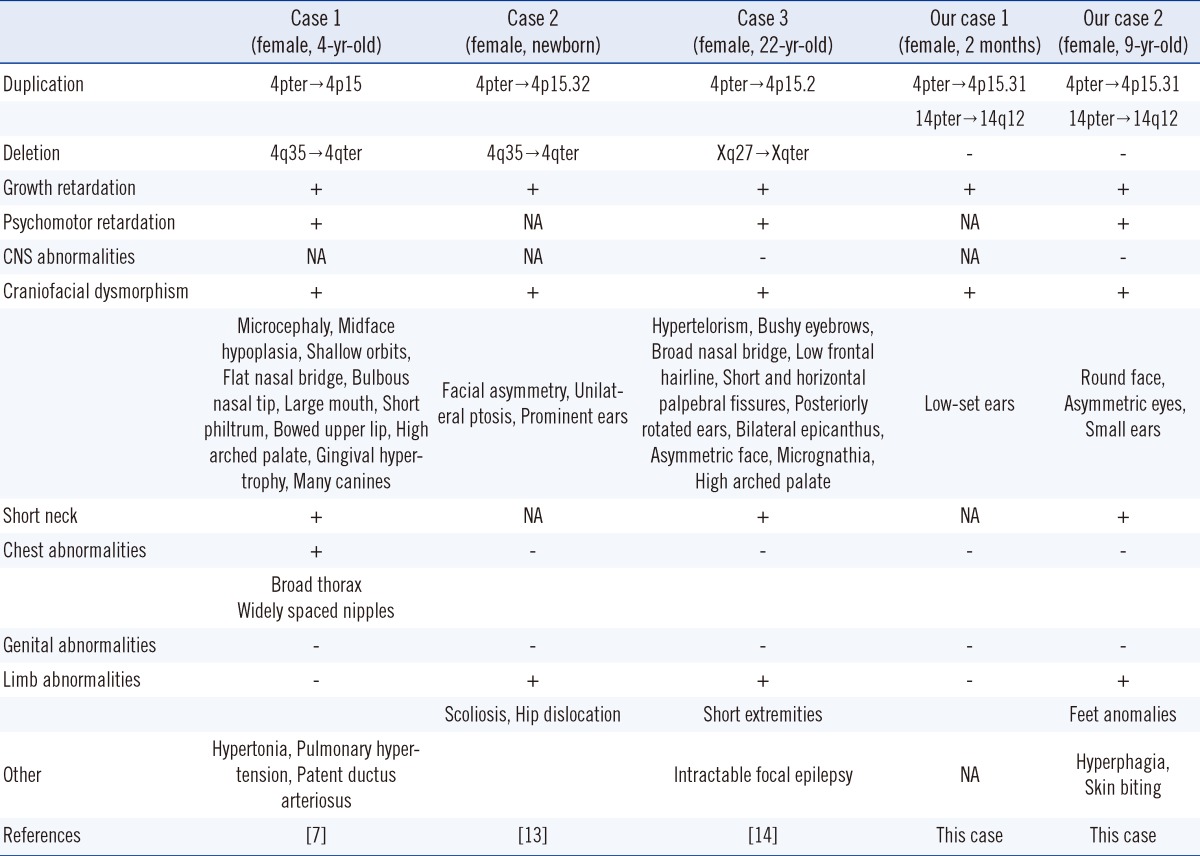
Comparison of the phenotypic characteristics of our patients with those of previously reported partial trisomy 4p or partial trisomy 14q cases revealed that the profound clinical manifestations of our patients were postnatal growth retardation and psychomotor delay, although the patient described in case 1 was too young to assess mental retardation (Table 1). In case 2, growth retardation was more prominent than weight gain. Other phenotypic abnormalities observed in both patients were, as in previous reports, variable but seemed to be milder. Considering that the phenotype of trisomy is generally milder than that of monosomy, the symptoms in our cases, which were 2 partial trisomies, would be expected to be milder than those in previous cases since the latter were accompanied by monosomy of the partner chromosome.
According to the results from the array analyses, the breakpoints in both cases were very similar even though these were 2 independent and unrelated families. Despite an apparently identical breakpoint, the phenotypes of the 2 cases showed distinct differences, such as the short palpebral fissure and low-set ears in case 1 and the round face, asymmetric eyes, small ears, short neck, finger/toe abnormalities, and behavioral problems in case 2 (Table 1). Comparison with previous partial trisomy 4p cases with similar breakpoints (4p15) also revealed that the spectrum of phenotypes was highly variable and inconsistent (Table 2) [7, 13, 14]. These phenotypic variances among cases with similar breakpoints could result from more detailed differences in duplicated segment size or from differences in other factors affecting phenotypic expression.
Despite the phenotypic differences, the experience from case 2 could be helpful for managing and counseling the younger patient (case 1). In particular, the strong response to growth hormone therapy, which led to 4 cm of growth after only 4 months of therapy in case 2, could be very informative for developing a treatment strategy for the patient described in case 1 as well as for patients with similar chromosomal abnormalities.
Interestingly, the behavioral problems observed in case 2, overeating and skin biting, are very unusual findings in partial 4p or 14q patients. Lemire et al. reported a partial trisomy 14q case with Prader-Willi-like characteristics, such as overeating, obesity, impulsivity, and compulsive skin picking [11]. However, the authors couldn't find any evidence of Prader-Willi syndrome in their cytogenetic and molecular studies, and they suggested that the presence of trisomy 9p could cause these unusual characteristics. Although our patient did not exhibit Prader-Willi characteristics other than overeating and skin biting, the probability that these behavioral symptoms are related to the 14q trisomy should be considered in future.
In conclusion, although our 2 patients had similar duplicated segments in 4p/14q, their phenotypes were not identical. Despite the fact that long-term follow-up is needed, growth retardation and developmental delay are likely to be the most notable findings in cases of combined partial trisomy 4p and 14q. In addition, careful examination is needed for cases with newly-identified, small, acrocentric chromosomes due to the possibility of a derivative chromosome.

 XML Download
XML Download