

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Apart from thyroid cancer, breast cancer is the most common cancer among Korean women [1]. Genetic alterations associated with breast carcinogenesis, including specific gene amplifications, deletions, point mutations, chromosome rearrangements, and aneuploidy, are well understood. In addition to these highly characterized mutations, epigenetic alterations are key contributors to breast carcinogenesis [2-6]. The most widely studied epigenetic event in breast cancer is hypermethylation of CpG islands associated with the promoter regions of several genes [7, 8]. Hypermethylation of CpG islands in gene promoter regions is thought to be especially relevant for the silencing of important growth control genes. For breast cancer, some of the genes reported to undergo hypermethylation are involved in evasion of apoptosis (DAPK, TWIST1, and HOXA5), cell cycle regulation (CDKN2A and CCND2), cell invasion and metastasis (CDH1 and APC), DNA repair (BRCA1 and GSTP1), and cell signaling (ER and RARb2) [2, 9]. These epigenetic alterations occur at an early stage in breast carcinogenesis.
However, most epigenetic studies have been performed on women in western countries, and the reported frequencies and disease specificities vary across the studies [3, 10-14]. These discrepancies most likely relate to differences in the populations studied, the methods used, and the genes and regions where methylation has been studied. Most of the methods used to detect methylation are labor-intensive and/or allow the study of the methylation status of only one gene at a time. In this study, we used an approach that allows the simultaneous assessment of aberrant promoter methylation of 24 tumor suppressor genes in normal and cancerous tissues from 60 Korean patients with primary breast cancer. The aims of this study were as follows: 1) to measure the frequency of gene hypermethylation in cancerous tissue and their corresponding normal tissue from Korean breast cancer patients and 2) to determine whether methylation changes in cancerous tissues from Korean breast cancer patients are associated with major clinicopathological features.
Go to : 
METHODS
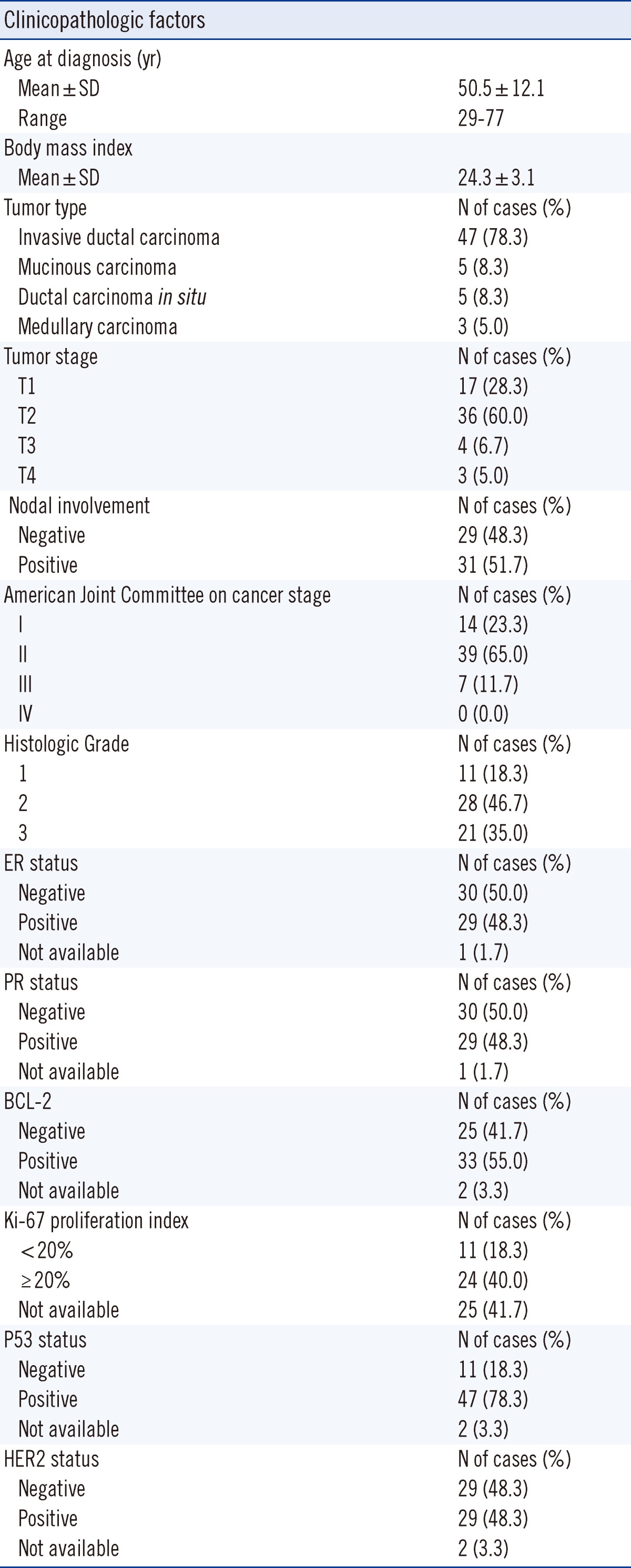
1. Patients and tissue samples
This study was approved by the Institutional Review Board of the Gyeongsang National University Hospital (Jinju, Korea), and written informed consent was obtained from all patients participating in the study. We consecutively collected 60 pairs of cancerous and the corresponding normal tissue samples from patients with breast cancer. All samples were procured at the time of surgery, subjected to an initial gross pathological examination, frozen at -180℃ in liquid nitrogen and stored until use. Corresponding normal tissues were procured from the most distant site from the resected specimen. For each tumor and normal breast tissue sample, a section adjacent to the tissue used for DNA extraction was stained with hematoxylin and eosin for histological confirmation of the presence or absence of cancer cells. However, tissue sizes were inadequate to perform tissue morphometry on these slides. Clinicopathologic characteristics of breast cancer were recorded by review of pathological files and electronic medical records (Table 1). Tumors were histologically graded from 1 to 3 according to the Nottingham modification of the Bloom and Richardson histological grading scheme [15]. Estrogen receptor (ER), progesterone receptor (PR), and p53 status were determined by immunohistochemistry. HER2 status was determined according to the College of American Pathologists (CAP) and the American Society of Clinical Oncology (ASCO) joint guidelines [16]. The mean age of the patients was 50.5 yr (range, 29-77 yr), and the most common tumor type was invasive ductal carcinoma (78.3%). More than half (51.7%) of the patients had pathologically positive nodes. In terms of biological markers, the ER and PR expression, Ki-67 proliferation index, HER2 status, and p53 overexpression were included in the analyses.
2. Tissue DNA extraction
Matched normal and tumor tissues were processed separately to avoid possible contamination. Each sample was frozen at -180℃ and broken with a frozen mortar. The homogenate was collected and resuspended in TE buffer (10 mM Tris/HCl and 1 mM EDTA). DNA was extracted by proteinase K digestion, followed by heat inactivation at 95℃ and phenol-chloroform and ethanol precipitation. DNA was quantified using a Nanodrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
3. Methylation-specific, multiplex ligation-dependent probe amplification assay
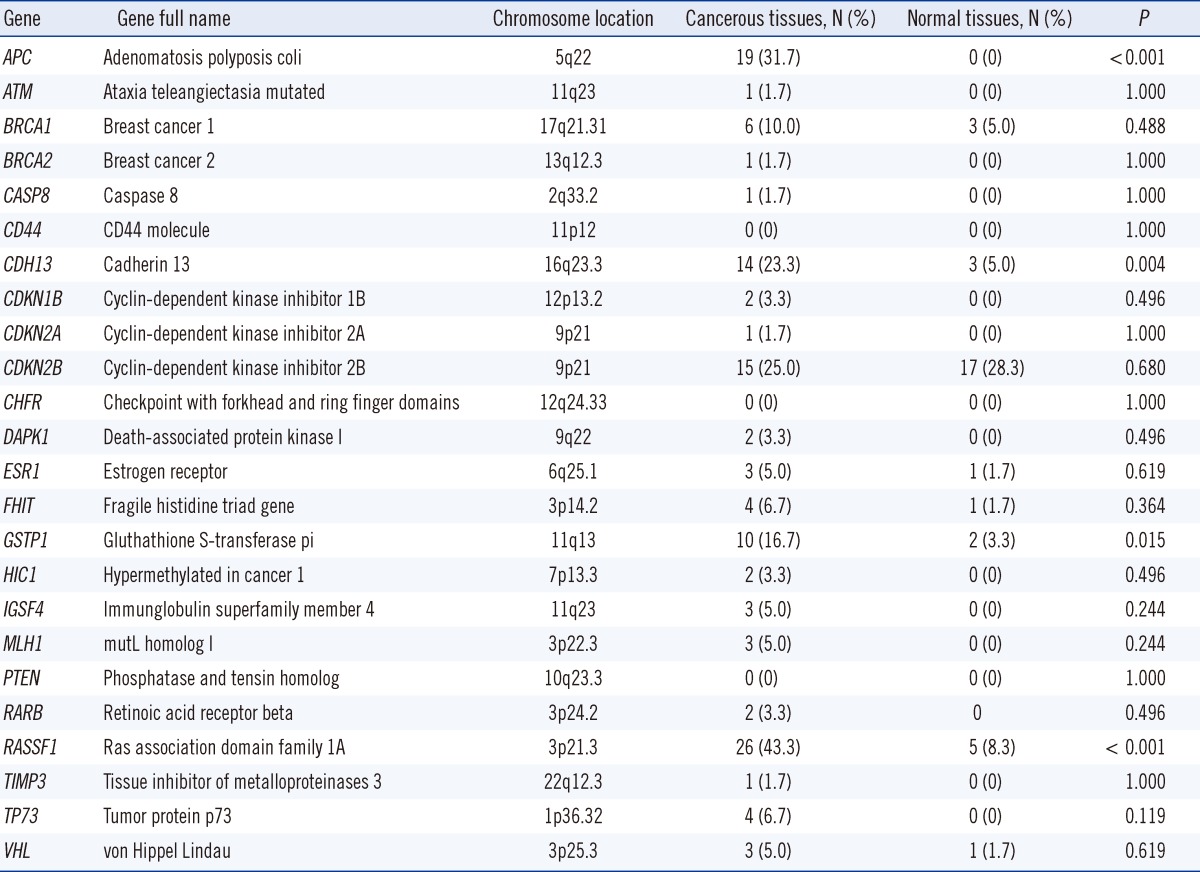
Gene methylation status was evaluated by methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) [17] by using the ME001-C1 Tumor suppressor-1 kit (MRC-Holland, Amsterdam, Netherlands). In all, 24 tumor suppressor genes and 12 internal control genes were studied using 50-100 ng of sample DNA (Table 2). In brief, during the first step of the assay, DNA samples were denatured and hybridized with target-specific MLPA probes by overnight incubation. Then, the reaction was split into 2 tubes. One tube was processed as the standard MLPA reaction, and the other tube was incubated with 10 U of HhaI at 49℃ for 30 min. Digested probes are not amplified by PCR and, hence, do not generate a signal. In contrast, if the target DNA is methylated, the hemi-methylated probe/sample DNA hybrids are prevented from digestion by HhaI and the target region is amplified, generating a signal. The amplified products were analyzed by sequence-type capillary electrophoresis (ABI 3100; Applied Biosystems, Foster City, CA, USA). The methylation status was subsequently quantified by comparing the relative signal peaks from the 2 tubes by using the GeneMarker_V1.90 software (SoftGenetics, State College, PA, USA). Methylation was scored as positive when the calculated methylation levels were higher than 25%. Any value below that level was considered negative.
4. Sample size calculation and statistical analyses
Van der Auwera et al. [18] demonstrated a 58.9% difference in RASSF1A gene hypermethylation between cancerous and morphologically normal tissues (76.8% vs. 17.9%, respectively). Thus, it was estimated that 41 patients per group would be required to provide a power of 95% to detect a statistically significant difference between the cancerous and morphologically normal tissues in the Korean patients with breast cancer by using a two-sided α-level of 0.05.
Continuous variables were presented as means±SD in baseline characteristics of patients with breast cancer (Table 1). For comparison of the methylation status of the 24 tumor suppressor genes in cancerous and corresponding normal tissues, data were presented as numbers and frequencies for categorical variables. The frequencies between groups were compared using the χ2-test or, in the case of low frequency per variable, Fisher's exact method. The Kappa statistic was used to assess the agreement between dichotomous variables between cancerous and corresponding normal breast tissue samples. The clinicopathologic parameters using categorical variables and the hypermethylation status in cancerous tissues in patients with breast cancer were analyzed using the Pearson's χ2-test or Fisher's exact method. A multivariate logistic regression analysis was used to identify independent predictors of clinicopathologic characteristics among the hypermethylated genes. Factors entered into the multivariate model were those with a P value less than 0.10 from the univariate analysis. A P value ≤0.05 of two-sided test was considered statistically significant. All statistical calculations were performed using SPSS, version 13.0 (SPSS Inc., Chicago, IL, USA).
Go to :
RESULTS
1. Methylation pattern of the 24 tumor-suppressor genes in cancerous tissue and their corresponding, morphologically normal breast tissue from patients with breast cancer
The distribution of DNA methylation of 24 tumor-suppressor genes in normal and cancerous breast tissues is shown in Fig. 1. Twenty one of 24 genes (87.5%) displayed promoter hypermethylation in one or more of the cancerous tissues (Fig. 1A). However, only 8 of 24 tumor-suppressor genes (33.3%) displayed promoter hypermethylation in the normal tissue samples (Fig. 1B). In the cancerous breast tissues, the most frequently hypermethylated genes were RASSF1 (43.3%) followed by APC (31.7%), CDKN2B (25.0%), CDH13 (23.3%), GSTP1 (16.7%), and BRCA1 (10%). Methylation was not observed in the CD44, CHFR, and PTEN gene promoters in cancerous tissues. In the normal breast tissues, the most frequently hypermethylated genes were CDKN2B (28.3%) and RASSF1 (8.3%). In their cancerous tissues, the frequencies of RASSF1 (43.3% vs. 8.3%), APC (31.7% vs. 0%), and CDH13 (23.3% and 5.0%) were significantly higher than those of normal tissues (P<0.001, P<0.001, and P=0.004, respectively; Table 2). However, the frequency of CDKN2B (25.0%) in cancerous tissues was similar to that (28.3%) in corresponding normal tissue (P=0.680).
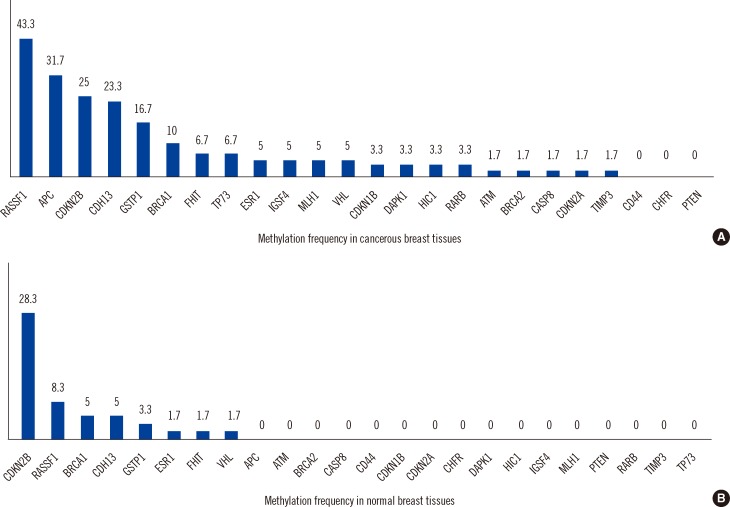 | Fig. 1Methylation frequencies of the tumor-suppressor genes in cancerous tissues and their corresponding normal tissues from the patients with breast cancer. (A) Methylation frequency of the 24 analyzed cancer-related gene regions in cancerous breast tissues. (B) Methylation frequency of the 24 analyzed cancer-related gene regions in the corresponding normal breast tissues. Numbers above each bar indicate the frequency of samples that were methylated in that region. The RASSF1 promoter was the most frequently methylated in cancerous tissues, and CDKN2B was the most frequently methylated in normal tissues. There were some gene regions whose methylation was not associated with breast tumor development (e.g., CD44, CHFR, and PTEN in tumors).
|
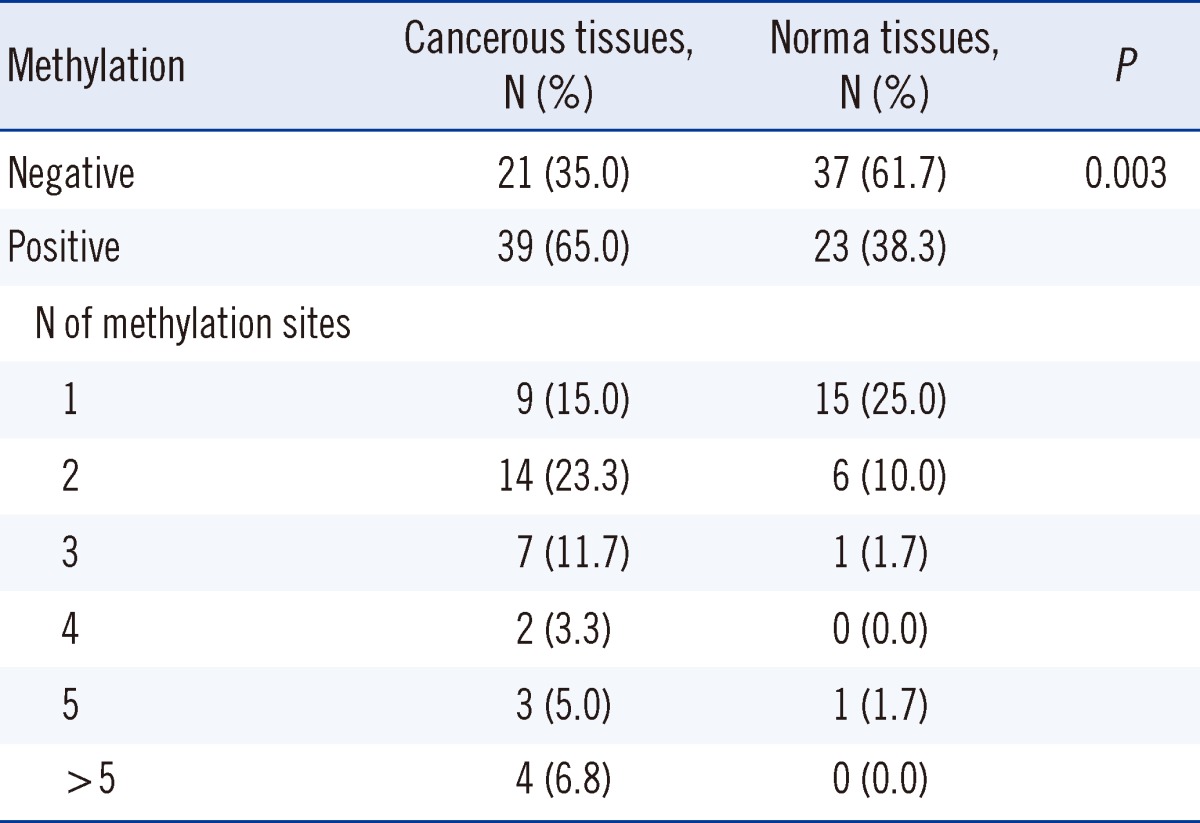
In 65% of the cancerous tissues, the promoter regions were hypermethylated in at least one of the 21 hypermethylated genes. The results are summarized in Table 3. One tumor sample was methylated at 14 sites. In 38.3% of the normal tissues, the promoter regions were hypermethylated in at least 1 of the 8 hypermethylated genes (Table 1). None of the normal tumor samples were methylated at more than 5 sites.
2. Association between DNA hypermethylation and clinicopathological factors
We investigated the association of clinicopathological features of the patients with the 7 most frequently hypermethylated genes (RASSF1, APC, CDKN2B, CDH13, GSTP1, BRCA1, and FHIT) in cancerous tissues. Table 4 shows the association between the hypermethylation of these 7 genes and various clinicopathological features of the patients. RASSF1 hypermethylation showed a statistically significant association with the invasive ductal carcinoma tumor type vs. other tumor types (53.2% [25/47] vs. 7.7% [1/13], P=0.032); however, post-hoc comparisons among these groups demonstrated no statistical significance between tumor types and RASSF1 hypermethylation. In addition, RASSF1 hypermethylation was associated with lymph node involvement (55.2% [16/29] vs. 32.2% [10/31], P=0.073), but the association was statistically insignificant. APC hypermethylation was associated with lymph node involvement (68.4% [13/29] vs. 19.3% [6/31], P=0.034). CDKN2B, CDH13, and GSTP1 hypermethylation were not associated with clinicopathological features. BRCA1 hypermethylation was associated with histologic grade III (25.0% [5/20] vs. 2.5% [1/40], P=0.013), negative ER expression (24.0% [6/25] vs. 0% [0/34], P=0.004), negative PR expression (20% [6/30] vs. 0% [0/29], P=0.014), and negative BCL-2 expression (20.8% [5/24] vs. 2.9% [1/34], P=0.046). The difference between the clinicopathological characteristics and BRCA1 hypermethylation in cancerous tissue was statistically significant as per the multivariate logistic regression analysis. FHIT hypermethylation was associated with lymph node involvement (13.8% [4/29] vs. 0% [0/31], P=0.049).
We also investigated the 5 most frequently hypermethylated genes, CDKN2B, RASSF1, BRCA1, CDH13, and GSTP1, in the corresponding normal tissues and examined their association with clinicopathological features. CDKN2B hypermethylation was associated with positive ER expression (40.0% [12/30] vs. 13.8% [4/29], P=0.032) and positive PR expression (37.9% [11/29] vs. 16.7% [5/30], P=0.055). Hypermethylation of RASSF1, BRCA1, CDH13, and GSTP1 in the corresponding normal tissues was not associated with clinicopathological features (data not shown).
3. Concordant gene methylation in cancerous tissue and corresponding normal breast tissue
For most of the 24 tumor suppressor genes, there was a poor to slight agreement between methylation in cancerous and corresponding normal tissues. The results of the concordant methylation pattern and disconcordant methylation pattern in the cancerous and corresponding normal tissues are shown in Fig. 2A, B, respectively. When at least one gene(s) was found to be hypermethylated in normal and cancerous tissues, poor agreement was noted (data not shown).
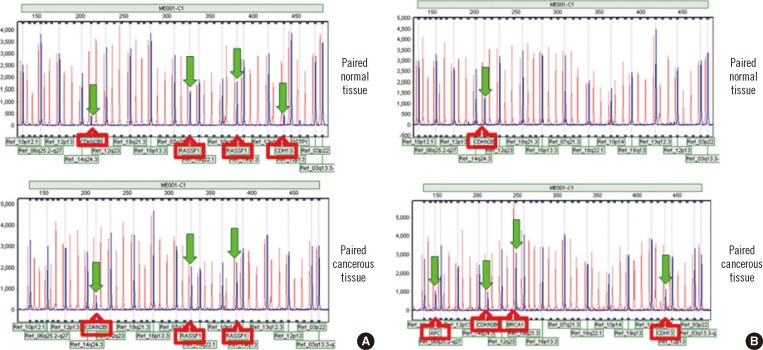 | Fig. 2Concordant vs. disconcordant methylation patterns in cancerous tissues and their corresponding normal tissues from the patients with breast cancer. (A) Concordant methylation pattern in normal and cancerous tissues from patients with breast cancer. Both tissues showed methylation changes for CDKN2B and RASSF1 genes. (B) Disconcordant methylation pattern in normal and cancerous tissues from patients with breast cancer. Morphologically normal tissue showed only CDKN2B methylation; however, the cancerous tissue showed methylation of multiple tumor suppressor genes, including APC, CDKN2B, BRCA1, and CDH13.
|
Go to :
DISCUSSION
Hypermethylation of CpG islands is a common mechanism for silencing tumor suppressor genes and occurs frequently in breast cancer. Although there is a large body of literature and the PubMeth online database (www.pubmeth.org) reporting tumor suppressor gene methylation frequency in breast cancer, methylation research of breast cancer in the Korean patients is still lacking, except for a few reports [5, 19, 20]. Several techniques exist to study aberrant methylation, i.e., methyl-specific PCR, bisulfite-dependent sequencing, and methyl-sensitive restriction enzyme-based assays. Most of these approaches are restricted to a limited number of genes. Although recent studies demonstrated that MS-MLPA analysis in breast cancer is relatively simple, sensitive, and highly specific to simultaneously detect the methylation status of multiple gene regions [17, 21-23], it is still necessary to confirm whether the MS-MLPA technique is an appropriate method to detect methylation.
In the present study, we investigated aberrant methylation of 24 genes in cancerous and their normal tissues from 60 Korean patients with breast cancer by using MS-MLPA. In the cancerous breast tissues, the most frequently hypermethylated genes were RASSF1 (43.3%), APC (31.7%), CDKN2B (25.0%), and CDH13 (23.3%). Using a PubMeth online database, which contains information and statistics about DNA hypermethylation in various cancers, and a review of the literature, the frequency of RASSF1 hypermethylation of breast cancer was determined to be 72.0% [22] and 71.4% [4], respectively. In this study, the frequency of RASSF1 hypermethylation was 43.34% (26/60) in cancerous tissues of all types of breast cancer and 53.2% (25/47), in cancerous tissues of invasive ductal carcinoma. This value is somewhat lower than the frequencies reported in previous studies and the online database [4, 24-26]. The discrepancy between these data might be the cut-off of positivity and analysis method rather than ethnic differences. However, these results are in accordance with the study by Buyru et al. [22], who reported that RASSF1 is the most frequently methylated gene and is methylated in 56.4% of invasive ductal carcinomas using the same MS-MLPA method as in this study. However, in a recent study based on the Korean ethnicity, the methylation frequency of ductal carcinoma in situ was 89% by using MethyLight PCR analysis, which was determined as positive when the percentage of methylated reference was >4 [19]. Therefore, the method and cut-offs of positivity of methylation analyses are very important to reduce false positives due to inadequate conversion of non-methylated cytosine to uracil and mis-priming when high numbers of PCR cycles or nested primers are used [5, 6]. Optimal cut-off determination can result in over-fitting and false-discovery. In the future, we will validate our findings by using a larger sample set from clinical trials and develop biomarkers for clinical use. RASSF1 hypermethylation has suggested lymph node involvement, but the association between hypermethylation and nodal involvement was statistically insignificant (P=0.073). RASSF1 methylation has been previously shown to be associated with nodal metastasis in patients with breast cancer [27]. Further, the frequency of RASSF1 methylation was shown to be higher in various metastatic sites as compared to the primary tumors [28], suggesting the potential of RASSF1 hypermethylation as a predictor for lymph node metastasis in breast cancer.
CDKN2B was the most frequently hypermethylated gene in corresponding normal breast tissues and the third most, in cancerous tissues. The CDKN2B hypermethylation has been frequently noted in various cancers, such as leukemia, lymphoma, colorectal cancer, brain cancer, liver cancer, gastric cancer, multiple myeloma, ovarian cancer, and lung cancer [29-32]. In cancerous, para-cancerous, and non-cancerous tissues in hepatocellular carcinoma, CDKN2B hypermethylation occurred at frequencies of 50%, 40%, and 25%, respectively [33]. The frequencies of CDKN2B promoter methylation in corresponding normal and cancerous tissues in colorectal cancer were reported to be 6.8% (6/88) and 26.1% (23/88), respectively [34]. Both studies suggested that CDKN2B promoter methylation might be an early event in hepatocarcinogenesis or colorectal cancer [33, 34]. This study demonstrates that CDKN2B promoter methylation in corresponding normal tissues in breast cancer is a frequent event and CDKN2B hypermethylation may be an early event of breast carcinogenesis in Korean patients with breast cancer.
The APC gene is the second most hypermethylated gene in cancerous tissues and the frequency was 31.7% (19/60); hypermethylation of this gene was not observed in corresponding normal tissues. Similar reports indicated that hypermethylation of the APC promoter was detected in 18 of 50 (36%) primary breast cancers and in none of the 21 non-cancerous breast tissue samples [35]. These results indicate that APC hypermethylation is a cancer-specific change. In addition, APC hypermethylation was associated with lymph node involvement in this study (P=0.034). Therefore, APC hypermethylation might result in more aggressive behavior, which can potentially become a useful prognostic indicator warranting a more aggressive therapeutic approach.
BRCA1 hypermethylation was the third most hypermethylated gene (5%) in the corresponding normal breast tissue and the fifth most (10%) in cancerous tissues in this study. Although there was no statistical significance between the clinicopathologic characteristics and BRCA1 hypermethylation using multivariate logistic regression analysis, BRCA1 hypermethylation was associated with negative ER expression (P=0.004), negative PR expression (P=0.014), negative BCL-2 expression, and high histologic grade (P=0.013). Methylation in breast cancer has long been linked to hormone regulation, but this correlation has not been established yet. The status of both ER and PR are very important to help determine patients who would benefit the most from hormone therapy. BRCA1 hypermethylation is more frequent among breast cancer patients with negative ER and negative PR expression, and this study suggests that hypermethylation of the gene promoter in cancerous tissues may be associated with hormone expression.
Detection of promoter CpG island hypermethylation offers several advantages compared to other DNA alterations in cancer. These events may provide ideal biomarkers for molecular diagnosis and early detection of cancer.
In conclusion, we have clearly demonstrated that normal-appearing breast tissue from the Korean patients with breast cancer exhibited frequent aberrant DNA methylation changes. This study also suggests that gene methylation may be linked to various pathological features of breast cancer; however, this requires confirmation in a larger study group.
Go to :
 XML Download
XML Download