

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Axenfeld-Rieger syndrome (ARS) is an autosomal dominant disease that manifests as anomalies of the anterior segment of the eye and systemic abnormalities [1]. In the eye, this condition is characterized by varying degrees of anterior segment dysgenesis and is associated with a high risk of glaucoma. Other associated systemic issues include cardiovascular outflow tract malformations, craniofacial abnormalities, and pituitary abnormalities that can cause severe endocrinological sequelae [2].
Genetically, mutations in FOXC1 on chromosome 6p25 and PITX2 on chromosome 4q25 have been identified in ARS patients [3, 4]. The prevalence of FOXC1 or PITX2 mutations in the affected probands ranges from 40% to 70% [5-7]. FOXC1 mutations are associated with only eye involvement, while PITX2 mutations often result in systemic abnormalities in addition to eye issues. Although the causative mutations have yet to be identified, 13q14 and 16q24 are known to contain loci of interest. A pediatric patient who was compound heterozygous for mutations in CYP1B1 showed characteristic anterior chamber anomalies and had an umbilical hernia [8-10].
PITX2, a member of the paired class of homeodomain transcription factors, is expressed in the corneal endothelium, stroma, iris, ciliary body, and sclera, which all originate from the neural crest. This transcription factor is known to affect the development of the periocular mesenchyme [8, 11]. A coding region frameshift as well as nonsense and missense mutations are thought to compromise the ability of PITX2 to bind to DNA. Copy number losses due to large chromosome rearrangements, small intragenic deletions, and a rare duplication have also been reported [12]. Here, we report a Korean family with clinical features of ARS carrying a novel c.300_301delinsT mutation in the PITX2 gene.
CASE REPORT
1. Case 1 (proband)
A 4-yr-old girl was referred to our clinic because of uncontrolled intraocular pressure (IOP) under maximal tolerable medical therapy and corneal edema in both eyes. Her best-corrected visual acuity was 20/800 in both eyes. IOP as measured by Goldmann tonometry was 22 mm Hg (reference range: ≤21 mm Hg) in the right eye and 23 mm Hg (reference range: ≤21 mm Hg) in the left eye. The horizontal and vertical corneal diameter of both eyes was 9.5 mm and 10.0 mm, respectively. Posterior embryotoxon and a prominent Schwalbe's line were observed in both eyes. Iridocorneal adhesion and corectopia were also seen, and anterior insertion of the iris into the trabecular meshwork with prominent iris processes was observed by gonioscopic examination in both eyes. The optic disc was difficult to inspect because of corneal edema; the cup-to-disc (CD) ratio appeared to be 0.8 in both eyes. The patient had apparent microdontia.
2. Case 2 (mother)
The 35-yr-old mother of the patient had 20/20 best-corrected visual acuity in both eyes. IOP as measured by Goldmann tonometry was 14 mm Hg (reference range: ≤21 mm Hg) in both eyes. Her cornea was clear, but focal iris atrophy was observed in both eyes. A posterior embryotoxon, which is a prominent Schwalbe's line, and anterior insertion of the iris into the trabecular meshwork with prominent iris processes were observed by gonioscopic examination in both eyes. Fundus examination revealed a normal appearing optic disc with a CD ratio of 0.3 in the right eye and 0.4 in the left eye. This patient also had redundant periumbilical skin and noticeable microdontia. She had dentures due to microdontia.
3. Molecular genetic analysis of PITX2
Sequencing analyses for FOXC1 and PITX2 in the proband and a family study for PITX2 mutations were performed. The purpose of the study and the procedures to be used were explained to all patients, and informed consent was obtained. Genomic DNA was isolated from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA). All exons with flanking intronic regions were amplified using the PCR with primers designed by the authors (available on request). Sequencing was performed with the ABI Prism 3100xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) using the BigDye Terminator Cycle Sequencing-Ready Reaction Kit (Applied Biosystems). Sequences were analyzed using Sequencher software (version 4.10.1, Gene Codes Corp., Ann Arbor, MI, USA) and compared with the reference sequence for PITX2 (NM_000325.5) and FOXC1 (NM_001453.2).
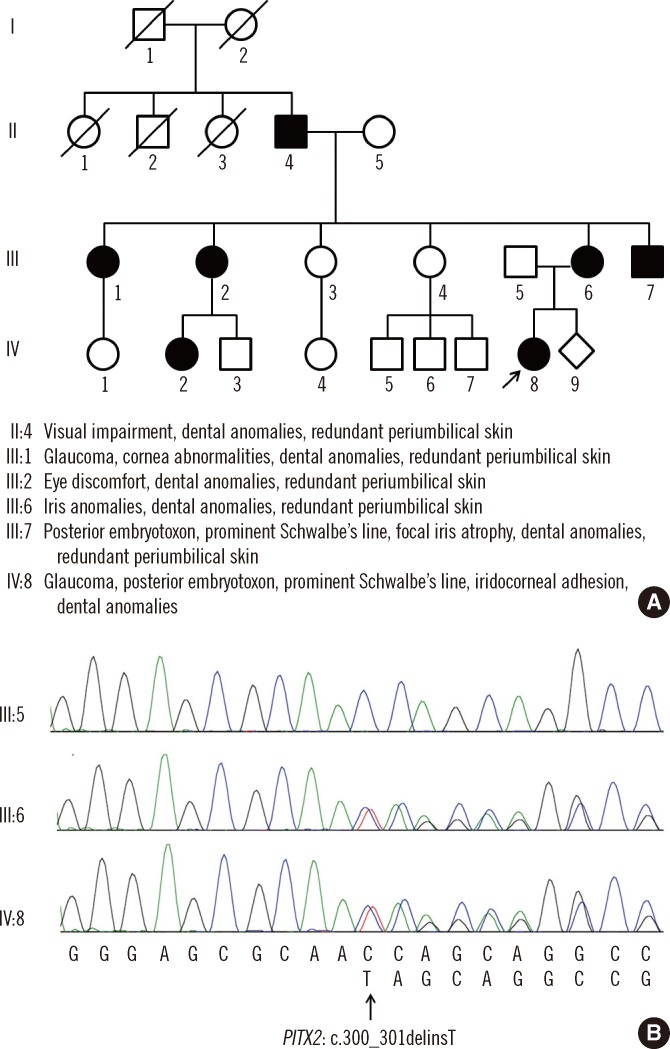
The patient was found to be heterozygous for a 2-bp deletion and an insertion of T in the PITX2 gene (c.300_301delinsT), a frameshift mutation predicted to result in premature termination at the 54th amino acid of the PITX2 protein (p.Gln101Serfs*54). Analysis of the parents revealed that this variant was inherited from the mother: the father had a wild-type sequence, while the mother had the same variant as her daughter (Fig. 1). When we tested 104 unaffected ethnically matched controls, none had the mutation. In addition, no other mutations were found in the PITX2 gene of the patient or her parents. Neither mutation nor unknown variation was found in the FOXC1 analysis.
DISCUSSION
We found a novel PITX2 gene mutation (c.300_301delinsT) that appears to result in ARS through haploinsufficiency either due to a frameshift-mediated decay of mutant mRNA or production of mutant PITX2 protein, in a Korean family affected by autosomal dominant ARS. The spectrum of Axenfeld-Rieger malformations demonstrates locus heterogeneity with the involvement of 2 major genes, PITX2 and FOXC1 [13]. This is the first report of a PITX2 gene mutation associated with ARS in a Korean individual. To our knowledge, 7 cases of ARS have been reported in Korea in addition to the present 2 cases. In one of those cases, the diagnosis of ARS was confirmed by a molecular method, which showed a novel FOXC1 mutation [14-17]. The clinical features, including ocular and extraocular symptoms and molecular features, are described in Table 1.
The PITX2 gene is located on 4q25, and mutations have been identified in 10-60% of probands with ARS [8, 18]. Most commonly, coding region frameshifts and nonsense and missense mutations are present, which are thought to affect DNA binding. A small number of large chromosome rearrangements, small intragenic deletions, and a rare duplication resulting in copy number loss have also been described [12]. All individuals who inherit the ARS-associated PITX2 or FOXC1 alleles exhibit ocular symptoms, with full penetrance [2]. Compared to FOXC1 mutations, PITX2 mutations are more commonly associated with the extraocular systemic abnormalities of ARS. In our case, the patient and her mother had extraocular abnormalities such as dental anomalies with or without redundant periumbilical skin. On the basis of the systemic findings, we predicted a PITX2 mutation rather than a FOXC1 mutation, but sequenced both genes. Although gain-of-function mutations in PITX2 have been suggested to cause ARS, most intragenic PITX2 mutations are loss-of-function mutations that yield a protein defective in DNA binding or one that is unable to transactivate downstream genes, or both [12]. FOXC1 transcriptional activity is negatively regulated by PITX2, which explains how FOXC1 duplication and PITX2 deletion result in similar phenotypes [19].
Phenotypes of the novel PITX2 mutation (c.300_301delinsT) in this family were variable. Although only the patient and her parents were fully genetically evaluated, ocular or extraocular manifestations of ARS were present in seven of the family members (Fig. 1). The patient showed severe glaucoma, Peters' anomaly, and visual impairment and therefore, requires a trabeculectomy and corneal transplantation. However, although her mother shared the PITX2 gene mutation, her ocular symptoms were mild and could be controlled with eye drops. In ARS, varying degrees of anterior segment dysgenesis may be present, and careful examination of the features of the anterior segment is necessary to establish the diagnosis for each family member. Other systemic symptoms including cardiovascular outflow tract malformations, craniofacial abnormalities, and pituitary abnormalities were not typical in this family. However, dental anomalies and redundant periumbilical skin were observed.
In summary, we found a novel c.300_301delinsT mutation in PITX2 in a Korean family with ARS. We suggest that this PITX2 mutation causes typical ARS. Our results extend the spectrum of PITX2 mutations and highlight the role of PITX2 in the development and progression of ARS.

 XML Download
XML Download