

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Familial juvenile hyperuricemic nephropathy (FJHN; OMIM 162000) is an autosomal dominant disorder characterized by a combination of hyperuricemia and gouty arthritis due to reduced kidney excretion of uric acid and progressive renal failure with renal tubulointerstitial fibrosis. Gradual progressive interstitial renal disease, with basement membrane thickening, and glomerulosclerosis resulting from fibrosis, starts in early life. Increases in serum creatinine generally occur between the age of 5 and 40 yr, resulting in occasional end-stage renal disease (ESRD) between the ages of 40 and 70 yr. There is some diversity in the age of onset of ESRD, both within and between families [1, 2].
FJHN was first reported in 1960, and only about 50 families of various ethnic origins have been reported [1]. Therefore, the prevalence and incidence of FJHN have not yet been established. FJHN is genetically heterogeneous [3, 4]; it is caused by mutations in the uromodulin gene (UMOD) located on chromosome 16p12.3.p13.11 [5], the HNF1 homeobox B gene (HNF-1B) on chromosome 17cen.q21.3 [6, 7], and an unidentified gene at chromosome 1q41 [8]. However, mutations in the UMOD are the most common changes responsible for FJHN. Mutations in UMOD are also associated with medullary cystic kidney disease type 2 (MCKD2; OMIM 603860), which is marked by corticomedullary cysts (not well documented in FJHN), tubulointerstitial inflammation, progressive renal failure, and variable hyperuricemia [3]; thus, MCKD2 and FJHN are considered to be allelic disorders. In addition, UMOD mutations have been described in glomerulocystic kidney disease (GCKD) [9, 10].
The UMOD consists of 11 exons, of which exons 2-11 encode uromodulin, also known as Tamm-Horsfall glycoprotein, which is an 85-kDa protein exclusively expressed in the kidney and is the most abundant protein in the urine of healthy individuals [11-13]. Uromodulin is produced exclusively in cells of the thick ascending limb and the early distal convoluted tubule, where it is expressed on the tubule lumen and excreted into the urine [14]. Patients with UMOD mutations excrete a markedly reduced amount of only wild-type uromodulin. Uromodulin comprises three epidermal growth factor (EGF)-like domains, two of which have Ca2+-binding motifs (cbEGF2 and cbEGF3), a zona pellucida domain, which is responsible for the polymerization of extracellular proteins into helical filaments and a glycosylphosphatidylinositol (GPI) anchor signal segment in the C-terminal region [15].
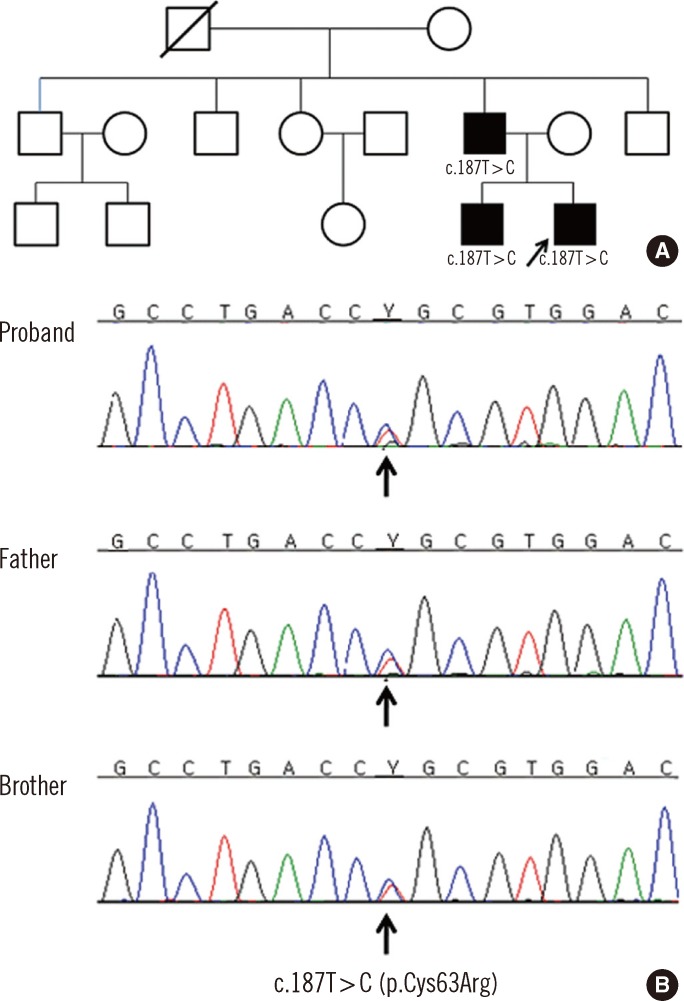
Thus far, only two FJHN cases have been reported in Korea, and only one of them was confirmed genetically. Here we report another Korean FJHN family with three affected male members-a father and two sons.and identify a novel UMOD mutation.
CASE REPORT
A 16-yr-old boy developed swelling and pain of the right malleolus about two weeks before hospitalization. He was evaluated at a local clinic and was treated with prednisolone under the diagnosis of hyperuricemia, gouty arthritis, and renal insufficiency. He was transferred to the department of nephrology at our hospital for further evaluation and proper management of abnormal renal function. At admission, his vital signs were stable and no symptoms related to azotemia were observed. Physical examinations, electrocardiography, and simple radiography did not reveal any abnormal findings. Blood urea nitrogen and serum creatinine levels were 36.9 and 2.12 mg/dL, respectively. Laboratory tests revealed hyperuricemia (uric acid, 10.3 mg/dL) and renal under-excretion of urate (24 hr urine uric acid, 9.6 g/day). Urine analysis showed mild hematuria (150 red blood cells/µL) and albuminuria (75 mg/dL). Other laboratory test results were within normal limits. Abdominal sonography showed normal size and echogenicity in both kidneys, with no other structural abnormality. Renal biopsy showed chronic parenchymal damage, coexisting with IgA nephropathy.
The proband had a family history of hyperuricemia, with his father and an elder brother having previously been diagnosed with hyperuricemia and gout. His father had received renal transplantation due to ESRD at the age of 34 in 1996. Considering the familial history, juvenile onset of hyperuricemia, gouty arthritis, and progressive renal impairment at early age, FJHN was highly suggested.
After obtaining written informed consent from all subjects, peripheral blood samples were obtained. Genomic DNA was extracted from peripheral blood leukocytes by using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA), following the manufacturer's instructions. Two exons (exons 3 and 4) of the UMOD and their flanking introns were amplified using primer sets designed by the authors (available upon request). The PCR was performed with a thermal cycler (model 9700, Applied Biosystems, Foster City, CA, USA) as follows: 32 cycles of denaturation at 94℃ for 30 sec, annealing at 60℃ for 30 sec, and extension at 72℃ for 30 sec. After treatment of the amplicon (5 µL) with 10 U shrimp alkaline phosphatase and 2 U exonuclease I (USB Corp., Cleveland, OH, USA), direct sequencing was performed with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems) on the ABI Prism 3130xl genetic analyzer (Applied Biosystems).
Sequence analysis revealed a novel heterozygous missense variation (c.187T>C; p.Cys63Arg) in exon 3 (Fig. 1). PolyPhen (Polymorphism Phenotyping, http://genetics.bwh.harvard.edu/pph2/) and SIFT (Sorting Tolerant from Intolerant, http://sift.jcvi.org/) programs predicted the p.Cys63Arg change to be "probably damaging" (PSIC score=3.410) and "affecting protein function," respectively. Both the father and an elder brother of the proband also carried the same variation. The variation identified in this family altered an evolutionary conserved residue in the UMOD.
The patient was treated with allopurinol (50-100 mg/day) and colchicine (0.6 mg/day) and his serum uric acid level began to decrease. Seven months later, his serum uric acid and serum creatinine levels were 8 mg/dL and 2.32 mg/dL, respectively. The elder brother was also treated with allopurinol and colchicine and both he and the proband have not developed ESRD to date and have remained clinically asymptomatic throughout this period.
DISCUSSION
The majority of families with FJHN have various heterozygous missense mutations in the UMOD located on chromosome 16p12.3. Mutations in UMOD have been identified in exon 3, 4, 5, and 7 [16]; however, more than 95% of those reported to date cluster in exons 3 and 4, which encode the three EGF-like domains, and a motif of eight cysteines within a cysteine-rich region [4, 16, 17]. In addition, more than 90% of UMOD mutations reported are missense substitutions, with the others being in frame deletions. Thus, all UMOD mutations are expected to result in a full-length protein; therefore, the resulting FJHN is presumed to be caused by abnormal protein folding, maturation, and trafficking, rather than uromodulin deficiency [16]. William et al. [16] demonstrated that UMOD mutations are associated with endoplasmic reticulum (ER) retention and delayed maturation and trafficking of abnormal uromodulin, which is probably due to protein misfolding. They also reported a possible association between the accumulation of mis-folded uromodulin within the ER and apoptosis through a stress-induced signaling pathway and resultant progressive tissue damage, which may be associated with the pathogenesis of FJHN due to UMOD mutations [15, 16].
Uromodulin is the most abundant protein in the urine of normal individuals. However, its biological role remains enigmatic, and it is unclear how UMOD mutations influence the reduced excretion of uric acid in patients with FJHN [18].
FJHN can be diagnosed by immunostaining of uromodulin, in which significantly modulated expression is detected [19] or by sequence analysis. However, immunostaining for uromodulin is not routinely performed by clinical laboratories and is only available at a limited number of institutes.
In the FJHN family described here, a heterozygote missense variant (c.187T>C; p.Cys63Arg) in exon 3 of UMOD was identified. We presume that this variant may be the causative mutation in this family, as it segregated with the disease. In addition, approximately two-thirds of known mutations lead to an alteration of a cysteine residue of uromodulin, and all such variants have been found to cause UMOD-associated kidney disease.
In conclusion, we report a Korean FJHN family with three affected members, identified by genetic analysis of the UMOD, and we provide the first report of a novel heterozygous missense mutation.
 XML Download
XML Download