

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Chronic granulomatous disease (CGD) is a rare genetic disease with an incidence of approximately 1 in 200,000 [1]. CGD is caused by a defect in the NADPH oxidase complex of phagocytes, which results in impaired production of superoxide anions and other reactive oxygen species (ROS), which are necessary for killing bacterial and fungal microorganisms. This defect leads to recurrent, life-threatening bacterial and fungal infections and granulomatous inflammation. Approximately two-third cases of CGD occur due to X-linked recessive mutations in the CYBB gene encoding gp91phox, which is located at Xp21.1. One-third of CGD cases occur as an autosomal recessive form, involving other genes encoding p22phox, p40phox, p47phox, and p67phox [2-4]. The dihydrorhodamine (DHR) assay is known to be a useful diagnostic tool for CGD [5]. We report a patient with X-linked CGD whose chimerism status following hematopoietic stem cell transplantation (HSCT) was rapidly determined using the DHR assay, which was compared to short tandem repeat PCR (STR-PCR) analysis.
Go to : 
CASE REPORT
A 3-month-old male infant presented with fever. His leukocyte count was 19.4×109/L; neutrophils, 12.086×109/L; Hb, 8.6 g/dL; platelet, 287×109/L; C-reactive protein, 6.57 mg/dL. Liver function tests showed AST activity of 158 U/L and ALT activity of 119 U/L. Computed tomography showed a hepatic mass in segment IV with a diameter of 50 mm, multiple satellite hepatic and splenic nodules, hepatosplenomegaly, and necrotizing mediastinal lymphadenopathy. Open liver biopsy revealed suppurative inflammation with abscess, and Serratia marcescens was identified from a culture of liver tissue. A liver abscess with S. marcescens and necrotizing mediastinal lymphadenopathy suspected of disseminated tuberculosis due to BCG injection raised concerns for CGD. Subsequently, treatment with antibiotics as well as antifungal and antituberculous medications was initiated.
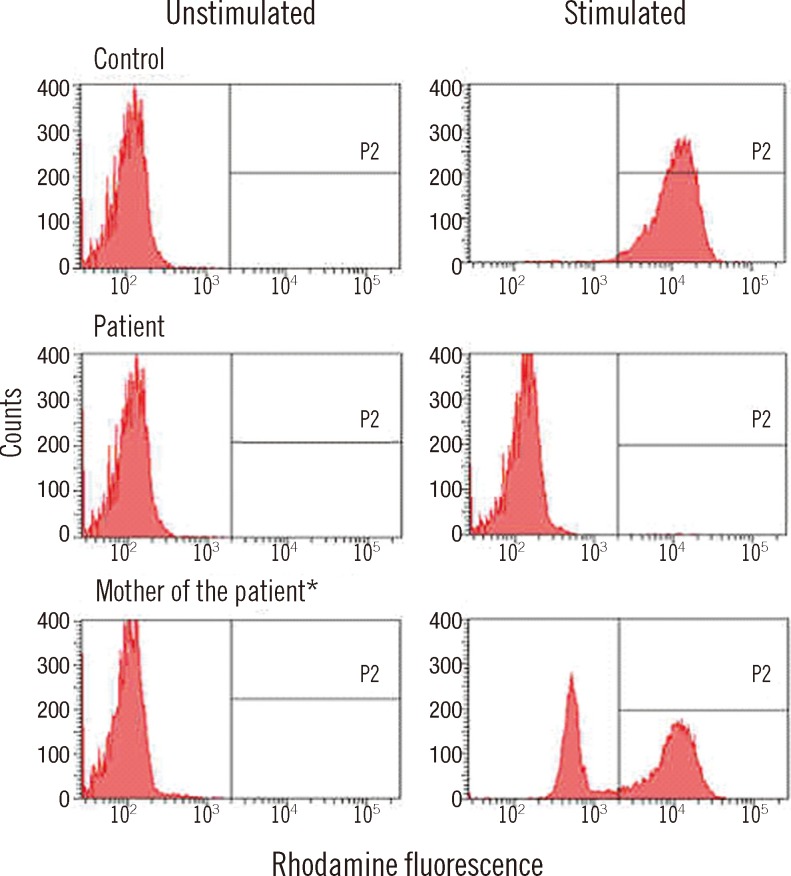
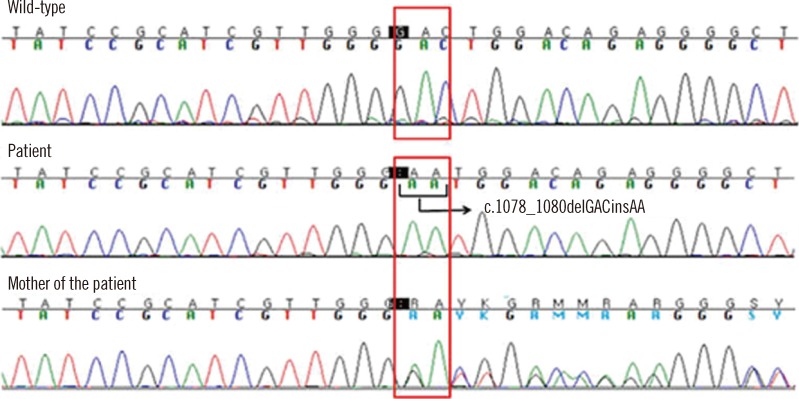
The neutrophil oxidative burst test was performed using the DHR assay before and after stimulation of neutrophils with phorbol myristate acetate (PMA) (Fig. 1). No DHR fluorescence was detected after stimulation (0.2%), consistent with a phenotype observed in X-linked CGD. Molecular genetic analysis of the CYBB gene encoding the gp91phox mutation, was performed using direct DNA sequencing to confirm the diagnosis of X-linked CGD. A novel frameshift mutation in exon 9 of the CYBB gene was identified (Fig. 2). The DHR assay and genetic tests performed on both the mother and brother of the patient revealed that the mother was a carrier of X-linked CGD but his elder brother was not. The family history was reviewed, and we found that the maternal uncle of the patient had died with pneumonia 2 months after birth, and he was, therefore, presumed to have been affected with CGD.
The patient received granulocyte transfusions 9 times at 4-5 months of age. Left hemihepatectomy was performed to remove a liver abscess during the same period; this surgery demonstrated chronic granulomatous inflammation. At the age of 7 months, multiple BCG granulomas, detected to be Mycobacterium tuberculosis by real-time PCR, were developed on the left upper arm as well as the neck and axillary area. Incision and drainage were subsequently performed.
At the age of 13 months, HLA-C 1 allele mismatched (11/12), allogeneic-unrelated, peripheral blood stem cell transplantation (uPBSCT) was performed under a conditioning regimen of busulfan (1.1 mg/kg), fludarabine (40 mg/m2), and rabbit anti-thymocyte (2.5 mg/kg). The blood group of the recipient and donor was A/Rh+ and B/Rh+, respectively. Dose of graft mononuclear cells, CD34+ cells, and CD3+ cells were 7.83×108/kg, 2.64×106/kg, and 2.38×108/kg, respectively. Engraftment of neutrophils and platelets occurred on day 13 and day 43, respectively, after HSCT.
Eighteen days after HSCT, preemptive treatment with ganciclovir for cytomegalovirus (CMV) viremia was administered. On day 27, amphotericin B treatment for probable invasive aspergillosis infection was initiated. On day 69 after HSCT, the patient developed grade II acute graft versus host disease of the gut.
Chimerism status following HSCT was monitored in peripheral blood or bone marrow mononuclear cells using STR-PCR analysis. At 2, 4, 8, 12, and 19 weeks after HSCT, donor cell chimerism was 68.3%, 94.1%, 84.7%, 40.9%, and 0%, respectively. The DHR assay, which was performed simultaneously, revealed 93.7%, 98.8%, 80.1%, 49.3%, and 0% of reactivity following PMA stimulation (Fig. 3). The fraction of the cell population, which previously showed a complete lack of DHR fluorescence, reappeared at 8 weeks after HSCT, and it gradually increased until defective neutrophils replaced the entire neutrophil population (Fig. 4). Finally, engraftment failure was determined by the absence of both donor cell chimerism and normal neutrophil population.
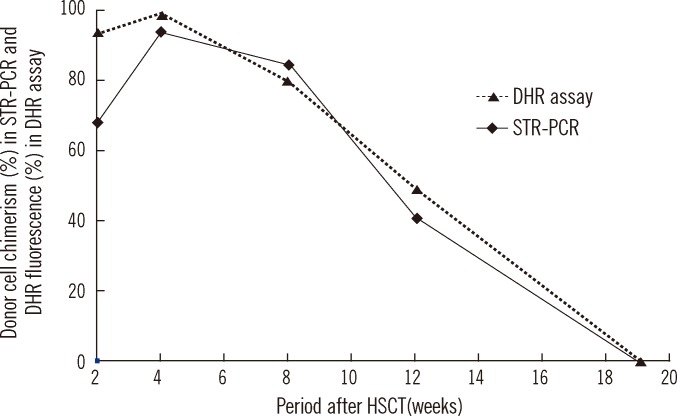 | Fig. 3Chimerism status in STR-PCR analysis and DHR assay during the follow-up period.
Abbreviations: HSCT, hematopoietic stem cell transplantation; STR-PCR, short tandem repeat polymerase chain reaction; DHR, dihydrorhodamine.
|
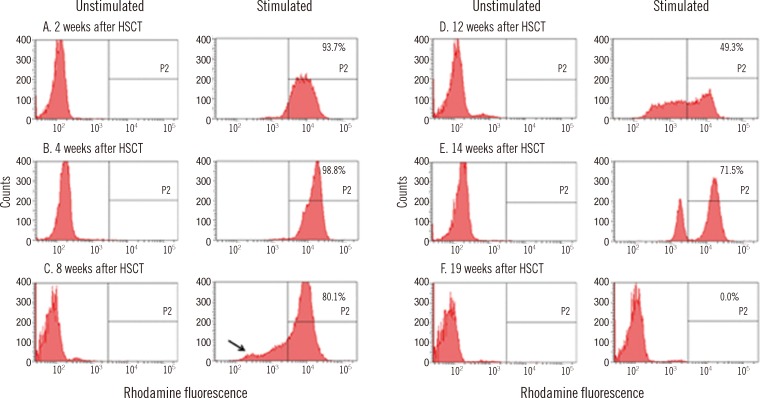 | Fig. 4DHR assay after HSCT. The black arrow indicates the small population showing complete lack of DHR fluorescence, which reappears 8 weeks after HSCT. The written number (%) is DHR fluorescence (P2) after PMA stimulation.
Abbreviations: HSCT, hematopoietic stem cell transplantation; DHR, dihydrorhodamine; PMA, phorbol myristate acetate.
|
The patient underwent a second HSCT from the same allogeneic donor after a conditioning regimen of total body irradiation (300 cGy), melphalan (45 mg/m2), and rabbit anti-thymocytes (2.5 mg/kg). Graft mononuclear cell, CD34+ cell, and CD3+ cell doses were 29.57×108/kg, 5.69×106/kg, and 18.53×108/kg, respectively. Engraftment of neutrophils and platelets occurred on day 14 and day 46 after HSCT, respectively. One hundred and twenty days after the second HSCT, the donor chimerism level monitored by STR-PCR was 100%, and defective neutrophil activity was not identified through DHR assay.
Go to :
DISCUSSION
CGD is diagnosed by an absence or reduction of oxidase activity in stimulated phagocytes, and it is confirmed by genetic testing. There are various tests for the measurement of neutrophil superoxide production, which include direct measurement of superoxide production, cytochrome c reduction assay, chemiluminescence, nitro blue tetrazolium reduction test, and the DHR assay. Of these tests, the DHR assay has proved to be a rapid and highly sensitive assay for CGD [6, 7]. This assay provides a clear discrimination between abnormal and normal phagocytes of CGD patients. Quantitative results of cellular responses are demonstrated as more or less impaired subgroups. DHR 123 freely enters the cell membrane, localizes in the mitochondria, and is oxidized to rhodamine 123 by ROS in stimulated phagocytes, which then emits a bright fluorescent signal that is measured by flow cytometry [5, 8]. The DHR assay can be helpful in the identification of the X-linked CGD and certain autosomal recessive type of CGD with a p47phox defect [7]. Detection of the X-linked CGD carrier is characterized by a mixture of normal and abnormal phagocytes [5]. Because the level of functional defect of neutrophils may differ among various genetic aberrations, especially in autosomal recessive CGD, the DHR assay is not always useful as a diagnostic method [9, 10]. Some exhausted neutrophils in a systemic inflammatory condition have been shown to have an attenuated response when activated in vitro [11].
There have been consistent advances in antibiotic and antifungal therapy and prophylaxis. However, CGD still causes significant morbidity and mortality, and the only known cure is successful allogeneic HSCT [12, 13]. Unrelated HSCT as well as related HSCT are performed more frequently, with improvement in overall survival rates. On the basis of recently reported outcomes, patient survival has increased from approximately 85% prior to 2000 to 90-95% in 2009 [12, 14]. A recent study by Martinez et al, [13] showed 100% survival for 11 patients undergoing HSCT, with a median follow-up of 2.5 years.
Analysis of the chimerism status after HSCT is important for evaluating engraftment and graft failure because early detection of imminent relapse provides a guide for intervention [15]. In many patients, the chimerism status is determined by STR-PCR analysis [16, 17]. This is performed through 2 different steps of amplification and capillary electrophoresis of short tandem repeats, and it has a sensitivity of 0.1-5×10-2 [17]. To use the STR-PCR for chimerism analysis, initial STR-PCR of the donor and pre-transplant recipient should be performed, and recipient informative alleles should be identified. Stutter peaks, an artifact of STR-PCR amplification, may develop, causing difficulty in distinguishing small recipient DNA peaks from stutter peaks [18]. STR-PCR analysis with DNA extracted from whole leukocytes has been shown to be incapable of distinguishing whether mixed chimerism in recipients is caused by the reappearance of normal recipient hematopoiesis or by the reoccurrence of malignant cells [19].
Although complete chimerism of donor cells is always desired, especially in malignant disorders, complete replacement of the recipient's hematopoietic system is not considered necessary to improve the underlying state of the disease in non-malignant disorders [20]. The degree of donor cell chimerism required for engraftment and correction of the underlying disease may be host- and disease-specific in patients with non-malignant disorders.
It has been reported that if there are greater than 10% of normal neutrophils, there is typically no evidence of a clinical phenotype in CGD [21]. Kuhns et al. [3] showed that the ROS production of phagocytes is a strong predictor of overall survival in CGD, and even small amounts of ROS production can have a significant survival benefit. In this patient, the DHR assay was conducted to monitor the recovery of phagocyte function after HSCT, and ROS production was observed as quantitative results. Additionally, quantitative results of the DHR assay correlated well with the donor cell chimerism status of STR-PCR. This finding shows the usefulness of the DHR assay as a monitoring tool supplementary to the PCR-based chimerism assay in post-HSCT X-linked CGD patients, in addition to highlighting its functional aspect as a monitoring tool of ROS production.
Recently, HSCT for CGD is becoming more common due to increased overall transplant success. In these circumstances, the DHR assay would facilitate the rapid determination of a patient's engraftment status, based on its advantages of simplicity, rapidity, and cost-effectiveness.
Go to :
 XML Download
XML Download