

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mucopolysaccharidosis III (MPS III), also known as Sanfilippo syndrome, is an autosomal recessive disease caused by a deficiency in 1 of the 4 enzymes that degrade heparan sulfate. MPS III includes 4 subtypes, types A to D, each of which results from a specific impaired lysosomal enzyme: heparan N-sulfatase (MPS IIIA, OMIM #252900), α-N-acetylglucosaminidase (NAGLU; MPS IIIB, OMIM #252920), acetyl-CoA:α-glucosaminide acetyltransferase (MPS IIIC, OMIM #252930), or N-acetylglucosamine 6-sulfatase (MPSIIID, OMIM #252940). The clinical features of MPS III primarily include central nervous system involvement and somatic abnormalities that are usually mild. The clinical course is divided into 3 phases [1]. The first phase, which usually occurs between 1 and 4 yr of age, consists of a developmental delay that often initially affects speech. The second phase generally starts at approximately 3-4 yr of age and is followed by severe behavioral problems, such as hyperactivity, challenging behavior, profound sleep disturbances, and progressive cognitive decline. The third phase, which usually occurs after the first decade of life, is associated with the onset of severe dementia. All motor functions start to decline, and the patient deteriorates into a vegetative state. Death usually occurs early in the third decade of life. However, there is considerable heterogeneity and longer survivals have been reported in patients with attenuated phenotypes [2, 3]. Clinical signs, symptoms, and the course of 4 subtypes of MPS III are indistinguishable.
MPS IIIB is caused by a deficiency of NAGLU. The NAGLU gene encoding α-N-acetylglucosaminidase was identified in 1996 and is located on chromosome 17q21.1 [4]. It contains 6 exons and encodes a 720-amino acid protein that has 6 potential N-glycosylation sites [5]. At present, over 120 different mutations in the NAGLU gene that result in MPS IIIB have been added to the Human Gene Mutation Database (HGMD, http://www.hgmd.org); these include missense/nonsense mutations, deletions, insertions, and splicing site mutations. Some cases of MPS IIIB have been reported in Korea; these cases were confirmed by an enzymatic activity assay of skin fibroblasts [6]. However, there have been no reports describing molecular genetic analyses of MPS IIIB. To our knowledge, this is the first study describing a MPS IIIB patient who was identified by performing biochemical and mutational analyses of the NAGLU gene in the Korean population.
CASE REPORT
A male patient was referred to our hospital at the age of 4 after he presented with mild coarse facial features and prominent broad eyebrows. There was no evident joint contracture. At the time of diagnosis, developmental delays were observed. Speech development was more delayed than motor development. His height (112.7 cm, 75-90 percentiles) and weight (23.5 kg, 95-97 percentiles) were within the normal range, as per the Korean growth charts. He was born at term with birth parameters in the normal range. The neonatal period was uneventful. The family had no history of metabolic disorders or any record of consanguinity. Upon clinical examination, no hepatosplenomegaly was found. Pectus carinatum and multiple persistent Mongolian spots were observed on his back. Whole spine x-ray radiography revealed ovoid-shaped lower thoracic vertebrae without other remarkable findings. Based on these clinical findings, screening tests for MPS, including cetylpyridinium chloride precipitation and toluidine blue tests, were performed. The biochemical detection of mucopolysacchariduria (657.2 mg glycosaminoglycan/g creatinine; reference range, <175 mg glycosaminoglycan/g creatinine) using the cetylpyridinium chloride precipitation test suggested the possibility of MPS. Thin layer chromatography revealed excessive urinary heparan sulfate. NAGLU enzyme activity was significantly decreased in leukocytes (not detected; reference range, 0.9-1.51 nmol/hr/mg protein) as well as in plasma (0.14 nmol/hr/mg protein; reference range, 22.3-60.9 nmol/hr/mg protein).
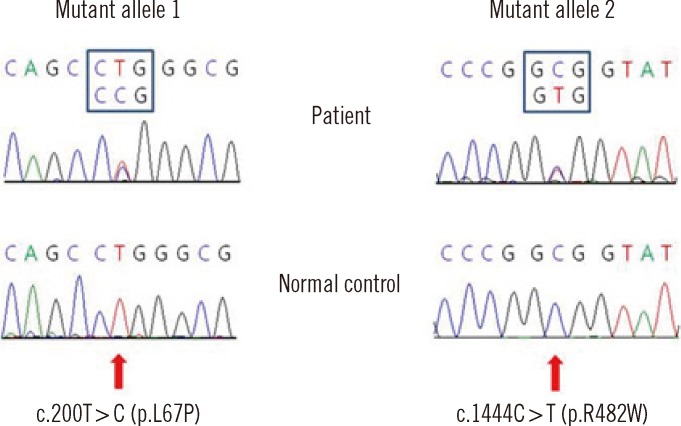
Blood specimens were collected from the patient after obtaining informed consent from his parents. Genomic DNA was extracted from the whole blood using the Wizard genomic DNA purification kit according to the manufacturer's instructions (Promega, Madison, WI, USA). All exons and intron-exon boundaries of the NAGLU gene were amplified. Direct sequencing of the PCR products was performed using the primers presented in Table 1. Cycle sequencing was performed using a Big Dye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, USA) on an ABI 3100 Genetic Analyzer (Applied Biosystems). The sequence obtained was compared with the reference sequence from the National Center for Biotechnology Information Database (http://www.ncbi.nlm.nih.gov). The nucleotides of the NAGLU cDNA were numbered according to respective GenBank accession numbers of NM_000263.3 for NAGLU. The Sorting Intolerant From Tolerant (SIFT) [7] and Polymorphism Phenotyping (PolyPhen-2 v2.1) [8] servers were used to predict the effects of a non-synonymous single nucleotide polymorphism (SNP) substitution on protein structure, function and phenotype, and sequence conservation. The patient was found to be a compound heterozygote for the missense mutations of c.200T>C and c.1444C>T. The latter mutation has been previously reported as a disease-causing mutation of MPS IIIB [9, 10]. The c.200T>C mutation is a novel mutation and induces an amino acid substitution of leucine to proline at codon 67 (p.L67P) (Fig. 1). Using different online bioinformatics softwares, the probability of amino acid alteration and its effects on protein function and pathogenicity were examined. SIFT 0 (≤0.05 pathogen) and PolyPhen-2 0.995 (0.5-1.5 possibly/probably damaging) were calculated for the amino acid substitution. The patient has no siblings, and a family study could not be conducted in this case. A control study conducted using exon 1-targeted sequence analyses for 100 alleles from 50 healthy subjects did not reveal any carriers of the c.200T>C mutation.
DISCUSSION
Early development of MPS III is typically normal, and its first signs are usually developmental delay and behavioral problems observed between the ages of 2 and 6. At initial diagnosis, speech development is generally much more delayed than motor development [11]. In this case, the patient had mild dysmorphic features with more delay in speech development than in motor development. These findings are consistent with MPS III. Little is known about the natural course of MPS IIIB, because few studies have been performed on the disorder. However, although there is high disease course variability, patients with MPS IIIB usually have a mild course [2].
The prevalence of MPS III has been reported to be 0.28-4.1 in Europe and 0.39 in Taiwan per 100,000 live births [11-14]. In MPS III, type A is the most common in northwest Europe, whereas type B is most frequent in southeast Europe, Taiwan, and Japan [10, 12, 15]. In Taiwan, the incidence of MPS IIIB was found to be 0.28 in 100,000 live births [12]. The incidence of MPS IIIB among Koreans is unknown. Sohn et al. [6] reported 7 MPS IIIB patients in Korea. However, there is a possibility that some patients with MPS III are misdiagnosed because of phenotypic variability and relatively mild somatic features. More than 120 mutations have been identified in the NAGLU gene, and 70.3% of them are missense mutations (HGMD, http://www.hgmd.org). In the present study, we identified 2 missense mutations: c. 200T>C (p.L67P) and c.1444C>T (p.R482W). The latter, p.R482W, has been previously reported in patients of Turkish and Japanese descent [9, 10]. It affects highly mutable CpG dinucleotides. The C to T mutation is in line with methylation-mediated deamination of 5-methylcytosine as one of the possible mutagenesis mechanisms. The p.R482W mutation in a Turkish patient was a homozygous mutation and was associated with a severe phenotype [9]. The novel mutation of c.200T>C (p.L67P) in the NAGLU gene was not found in 100 alleles from 50 healthy subjects. In addition, in silico prediction analyses using PolyPhen-2 and SIFT revealed the possibility of c.200T>C being a disease-causing mutation. Our patient showed significantly decreased NAGLU enzyme activity in leukocytes and plasma, which is consistent with the phenotype of MPS IIIB. Thus, the c.200T>C (p.L67P) mutation is thought to be a disease-causing mutation according to our control study, in silico analyses, and the clinical presentation of our patient.
MPS IIIB is known for its poor correlation between genotype and clinical phenotype. Allele frequencies of the different mutations are low, and the majority of mutations are unique to individual families. The allelic heterogeneity is assumed to contribute to the wide spectrum of clinical phenotypes [11, 16-18]. The prediction of genotype-phenotype relation in MPS IIIB is complicated by numerous polymorphisms that potentially modify disease severity [17]. A minority of patients with MPS IIIB has been reported with severe phenotypes; these patients showed mutations resulting in premature termination by nonsense mutations, insertions, or deletions [16, 17, 19]. However, p.R643C, p.S612G, p.E634K, and p.L497V have been reported to be associated with more attenuated phenotypes [2].
To the best of our knowledge, this is the first molecular genetic analysis of an MPS IIIB patient from the Korean population. We identified a novel mutation (c.200T>C) in the NAGLU gene. This report will contribute to a better understanding of the genetic background in Korean MPS IIIB patients. Further clinical, biochemical, and molecular studies are needed to establish better genotype-phenotype correlations in MPS IIIB mutations and symptoms.

 XML Download
XML Download