

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ataxia-telangiectasia (A-T; MIM #208900) is an autosomal recessive, multi-system disorder with onset during childhood that is characterized by progressive cerebellar ataxia, oculomotor apraxia, oculocutaneous telangiectasias, choreoathetosis, immunodeficiency, frequent infections, elevated alpha-fetoprotein (AFP) levels, chromosomal instability, and an increased risk of malignancy, particularly leukemia and lymphoma [1]. A-T cells are hypersensitive to ionizing radiation and radiomimetic chemicals because of defects in cell cycle checkpoints that are normally activated by DNA damage [2].
The ataxia-telangiectasia mutated (ATM) gene-localized on chromosome 11 at position q22-23.has been identified by positional cloning [3]. The gene encodes a large protein, ATM, with a molecular mass of approximately 350 kDa. The protein encoded by the ATM gene belongs to the phosphatidylinositol (PI)3/PI4 kinase family, which is a cell cycle checkpoint kinase. The protein functions as a regulator of a wide variety of downstream proteins, including tumor suppressor proteins p53 and BRCA1, checkpoint kinase CHK2, checkpoint proteins RAD17 and RAD9, and the DNA repair protein NBS1 [4]. Most ATM mutations are private mutations and, to date, no mutational hotspots have been identified in the ATM gene.
Although A-T is known in most countries to be the most common cause of progressive cerebellar ataxia in childhood, there have been no confirmed cases in Korea. In this report, we present the clinical features of a pair of Korean siblings with A-T that were found to be heterozygous for two ATM gene mutations.
Go to : 
CASE REPORT
1. Clinical findings
An 8-yr-old boy and his 6-yr-old sister presented to the neurology outpatient clinic with a chief complaint of progressive ataxia. The patients displayed progressive limb and truncal ataxia, as well as involuntary movements of the extremities. There was no history of perinatal complications, cognitive dysfunction, or seizures. The proband had developed recurrent respiratory infections. The parents were healthy, non-consanguineous ethnic Koreans. The proband was the first child in his family, and no family history other than the two patients was known.
The proband first presented to the pediatrics clinic four years prior to the neurology visit. A blood test at the initial visit showed mild anemia (11.6 g/dL), thrombocytosis (441×109/L), and an elevated phosphate level (5.2 mg/dL). Serum levels of lactate, pyruvate, and cholesterol were within normal limits. There were no detectable abnormalities in the urine, and the levels of serum amino acids and urine organic amino acids were normal. Left undiagnosed for three years, the patient had only received conservative care and rehabilitation.
When both siblings presented to the department of neurology, the proband exhibited more severe symptoms. A neurological examination revealed the presence of limb and truncal ataxia, oculomotor apraxia with gaze-evoked nystagmus, and choreoathetosis of the bilateral extremities. There was no cranial nerve dysfunction, motor weakness, or sensory dysfunction. Deep tendon reflexes were decreased. Ophthalmologic evaluation did not reveal telangiectasia in either of the children. The AFP level was increased (119.68 IU/mL) in the proband's serum. No chromosomal abnormalities were found. The number of CAG repeats was not increased in the genes for spinocerebellar ataxia type 1, 2, 3, 6, 7, 8, or 17. Cerebellar atrophy on brain magnetic resonance imaging (MRI) images taken two years prior to the visit was more prominent than that in the initial image taken 4 yr ago.
2. Molecular genetic analysis
After obtaining informed consent from the parents, the genomic DNA was isolated from peripheral blood leukocytes of the proband and his siblings by using a Wizard genomic DNA purification kit (Promega, Madison, WI, USA). All the coding exons and flanking intronic regions of the ATM gene were amplified using primer sets designed by the authors (sequences available upon request). PCR was performed using a thermal cycler (Model 9700; Applied Biosystems, Foster City, CA, USA). Direct sequencing was performed using an ABI Prism 3130xl Genetic Analyzer (Applied Biosystems) with a BigDye Terminator Cycle Sequencing-Ready Reaction Kit (Applied Biosystems).
RNA was extracted to perform reverse-transcription PCR (RT-PCR). cDNA was generated using an RNA PCR kit (Applied Biosystems). RT-PCR.based sequencing was performed using a set of primers (RT-F, 5'-CCAATTCTTCACAGTAATTTTCCTC-3'; RT-R, 5'-CGATACAAAGAACACACATTGGA-3'). The PCR product obtained was subjected to direct sequencing as described above.
The cDNA nucleotide sequences of the gene examined were numbered according to the GenBank accession number NM_000051.3 for ATM. The mutation nomenclature used here follows the recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/).
3. Molecular genetic findings
In the proband, a previously reported heterozygous mutation at position 8,546 (c.8546G>C; p.Arg2849Pro) [5] and a 13-base deletion in intron 17 (c.2639-19_2639-7del13) were identified (Fig. 1). RT-PCR analysis revealed that the intronic variant resulted in a marked increase of an aberrant transcript missing exon 18 (954 bp) that was only faintly observed in normal controls (Fig. 2). These two variants were also present in the affected sister, but they were absent in the proband's healthy brother. In addition, the c.2639-19_2639-7del13 variant was not found in 100 control chromosomes.
 | Fig. 1(A) Pedigree of the family with A-T and (B-C) genomic DNA sequence analysis of the ATM gene in the proband and his siblings. (B) The patient was identified as being compound heterozygous for mutations of the ATM gene. The solid arrow indicates overlapping peaks at nucleotide position 8,546 because of a heterozygous G>C transition (c.8546G>C, p.Arg2849Pro). (C) The open arrow indicates the presence of successive double peaks at nucleotide position 2,639-19, suggesting the deletion of 13 base pairs (c.2639-19_2639-7delGAGTGCTTTTTAT).
Abbreviations: A-T, ataxia-telangiectasia; ATM, ataxia-telangiectasia mutated.
|
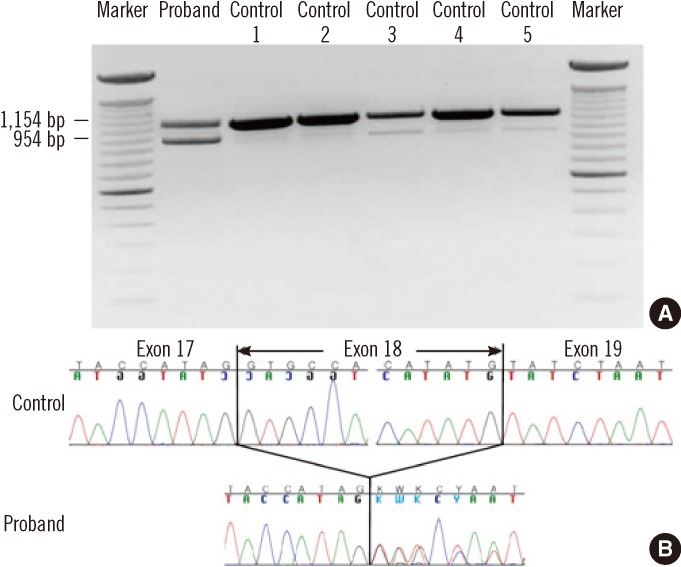 | Fig. 2RT-PCR sequencing of the ATM gene. (A) Electrophoresis of RT-PCR products using the primer pair described in the Case Report demonstrates an additional band (954 bp), as well as the expected band (1,154 bp), in the proband. The density of the aberrant band is much stronger in the proband than in controls. (B) Sequencing of the RT-PCR products demonstrated the aberrant skipping of exon 18.
Abbreviations: RT-PCR, reverse-transcription polymerase chain reaction; ATM, ataxia-telangiectasia mutated.
|
Go to :
DISCUSSION
A-T is a rare neurodegenerative disease, but in most countries, it is the most frequent autosomal recessive cause of progressive cerebellar ataxia in childhood [6]. A-T has a wide ethnic and geographic distribution. The prevalence of A-T has been reported to range from 1 in 40,000 to 1 in 300,000 live births, depending on the geographic or ethnic region [7, 8]. Moreover, the type and frequency of specific mutations in different localities can vary widely, with some regions having a specific founder mutation [9, 10]. In Asia, a nationwide survey was conducted to identify A-T patients in Japan, and the first reports of Chinese patients with ATM mutations surfaced in 2006 [11, 12]. The prevalence of A-T in the Korean population appears to be extremely low, with only a few cases reported thus far [13, 14]. The precise incidence, clinical characteristics, and genetics of A-T in the Korean population are unknown because of a lack of epidemiological surveys.
The patients in this study showed signs and symptoms of A-T, such as cerebellar ataxia, oculomotor apraxia, and elevated AFP levels. Contrary to the commonly associated characteristics, these patients' eyes and skin were without telangiectasias. Therefore, variant A-T-defined as the absence or presence later in life of some of the hallmarks of classic A-T.could not be ruled out, although telangiectasia may appear several years after the onset of neurologic symptoms [15-17].
On mutation analysis, our patients were shown to have two heterozygous variants. The c.8546G>C (p.Arg2849Pro) mutation, located in exon 58, is a known mutation that was first described by Sandoval et al. [5]. This mutation was reported to reside within the kinase domain and to affect residues that are highly conserved in the ATM-homologous proteins of different species. In addition, a novel c.2639-19_2639-7del13 variant in intron 17 was identified. RT-PCR analysis of the ATM mRNA showed that this variant resulted in aberrant skipping of exon 18. Although a small amount of alternatively spliced transcript without exon 18 was detected in normal controls, the amount of aberrant transcript markedly increased with the variant.
The clinical diagnosis of A-T is difficult when typical manifestations such as telangiectasia are absent. In addition, A-T is quite rare in Korea, which could lead to a delay in the diagnosis. Detecting ATM mutations in A-T patients could be useful diagnostically, especially in the early stages or in variant A-T. Although curative treatment strategies are not available for A-T, early diagnosis is of particular importance, given the increased risk of malignancy. The early recognition of A-T is a prerequisite to provide a correct prognosis and appropriate rehabilitation and support, including the avoidance of diagnostic X-ray procedures because of A-T.related radiosensitivity [16, 17].
Unfortunately, we could not confirm the compound heterozygosity of the 2 variants identified in this study, because both parents of the patients refused genetic testing for themselves, and the younger sibling of the patients did not have either of the two variants. Nevertheless, the presence of a novel splicing variant, as well as a known mutation in the two patients with A-T, highly suggests that the two variants might be causative mutations in the siblings.
In conclusion, this study reports Korean siblings with A-T carrying a known mutation and a novel splicing variant of the ATM gene. It should be noted that the diagnosis of A-T should be suspected, even in Korea, in patients exhibiting the clinical features of A-T. This study highlights the need for additional research on a large set of patients in this regional population. It is expected that such studies will help us better understand this serious disorder and develop appropriate therapeutic strategies.
Go to :
 XML Download
XML Download