

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
MDS is a heterogeneous group of clonal hematopoietic stem cell disorders characterized by bone marrow (BM) failure, dysplasia of peripheral blood and BM cells, and a high risk of transforming into an acute leukemic phase [1]. MDS occurs as a result of ongoing DNA damage, natural depletion of stem cells, and accumulated exposure of the BM to environmental stresses or toxins [2]. Thus, the risk of MDS increases with age. Childhood MDS is relatively rare compared to adult MDS [3]. The incidence of childhood MDS is less than 5% of all hematologic malignancies in childhood [4, 5].
Overall, approximately 30% of MDS progresses to acute leukemia, typically AML. The risk of transformation into AML differs among MDS subtypes according to WHO classification. In less than 5% and up to more than 50% of cases, MDS eventually progresses to AML after 5 yr [6]. However, progression of MDS to ALL is rare, occurring in less than 1% of adult cases, and even fewer in the pediatric population [7]. To our knowledge, there has been no case report of childhood MDS transforming into ALL in Korea, and there are only a few case reports worldwide [8-10]. Here, we report a rare case of an 8-yr-old Korean girl who presented with childhood MDS-refractory cytopenia that transformed into ALL 3 months after the initial diagnosis of MDS.
CASE REPORT
An 8-yr-old girl was transferred to our hospital for further evaluation of severe pancytopenia that was discovered 4 days earlier at a local hospital, to which she had been admitted for fever persisting for 2 weeks. Her hemoglobin was 6.0 g/dL, white blood cell (WBC) count was 0.9×109/L, and platelet count was 71×109/L. She had mild, easy bruising on both legs and night sweating. At admission, the patient had an acute ill appearance without fever and showed no splenomegaly, lymphadenopathy, or organomegaly on physical examination. A complete blood cell count (CBC) test showed hemoglobin of 5.8 g/dL, WBC count of 2.4×109/L, and platelet count of 214×109/L. A peripheral blood smear revealed severe normocytic normochromic anemia with mild anisocytosis and poikilocytosis, including some dacrocytes, moderate neutropenia (absolute neutrophil count of 0.78×109/L) with left shifting, and a few giant platelets. Her reticulocyte count was in the reference range (1.11%) and nucleated red blood cells (RBC) were observed (1/100 WBCs). Schistocytes were not observed. Most biochemical tests for liver and renal function tests were within the reference intervals, except increased serum lactate dehydrogenase (LDH; 638 IU/L, reference interval, 218-472 IU/L) and alkaline phosphatase (ALP; 506 IU/L, reference interval, 95-280 IU/L). Serum C-reactive protein (CRP) was slightly increased at 5.3 mg/L (reference interval, 0-5.0 mg/L). Urinalysis results were not specific except a few WBCs, 3-5/high-power field.
BM aspirate smears and touch print preparation of the BM biopsy revealed hypercellular marrow with dysplastic changes in 3 hematopoietic cell lineages. Erythroid dysplasia, including nuclear budding, multinuclearity, and nuclear irregularity, was present in 15% of all erythroid precursors (Fig. 1A). Granulocytic dysplastic changes, including megaloid change, hyposegmentation, and hypogranularity of the cytoplasm, was present in 15% of granulocytic cell lines. Megakaryocytes were adequate in number and exhibited dysplastic changes, including unequivocal micromegakaryocytes, separated nuclear lobes, and non-lobulated or round nuclei in 10% of all megakaryocytes. Myeloblasts accounted for 0.8% of nucleated cells in the BM. A BM biopsy section exhibited nearly 100% cellularity with a marked increase of erythroid precursor cells. Dysplastic changes in all hematopoietic cell lines were the same as in BM aspirates. Few histiocytes were observed in the biopsy section (Fig. 1B).
The pathologic findings of aplastic anemia or BM failure diseases such as adipocytosis and hypocellular or acellular marrow were not observed. Flow cytometric analysis for CD55 and CD59 performed to rule out paroxysmal nocturnal hemoglobinuria revealed normal levels of CD55 and CD59 in both RBC and WBC. Epstein-Barr virus real-time quantitative PCR and parvovirus B19 IgM tests revealed no evidence of viral infection. Nutritional deficiencies and metabolic diseases were excluded based on medical history and physical examination.
Cytogenetic analysis of the BM revealed a normal karyotype (46, XX). No cytogenetic abnormalities were detected in FISH analysis with probes for EGR1 (5q-), D7S486 (7q-), CEP8 (trisomy 8), and D20S108 (20q-). The patient was diagnosed as having refractory cytopenia of childhood (RCC) based on the 2008 WHO classification system. She received only supportive treatment in our hospital. After her general condition recovered, she was discharged and followed up with CBC and liver function tests.
Three months after the initial diagnosis of MDS, she was re-admitted to our hospital because of a relapse of high fever. A CBC test revealed hemoglobin of 9.4 g/dL, WBC count of 410.5×109/L, and platelet count of 15×109/L. A peripheral blood smear revealed severe microcytic hypochromic anemia, marked leukocytosis with many leukemic blasts (90%), and severe thrombocytopenia. The results of several biochemistry tests were increased as follows: AST, 134 IU/L; ALT, 74 IU/L; ALP, 498 IU/L; LDH, 11,306 IU/L; CRP, 54 mg/L.
BM aspirate smears (Fig. 2A) and a biopsy section (Fig. 2B) revealed a markedly hypercellular marrow that had been completely replaced by small leukemic blasts (95%). Normal hematopoietic cells were markedly decreased. Cytochemical staining demonstrated that the cells were all negative for myeloperoxidase (MPO), Sudan black B (SBB), and periodic acid-Schiff (PAS). Flow cytometric immunophenotyping revealed that the blasts expressed B lymphoid markers that were CD10 (+), CD19 (+), CD79a (+), cytoplasmic IgM (+), and terminal deoxynucleotidyl transferase (TdT) (+). The myeloid cell markers of CD13 and CD33 and T cell markers of CD2, CD5, and CD7 were not expressed. Karyotype analysis of BM leukemic cells revealed normal chromosomes (46, XX). BCR-ABL, E2A-PBX, and TEL-AML gene rearrangements were not detected using reverse transcription-PCR and FISH analyses. MLL gene rearrangements and p16 (9p21) deletion analysis by FISH were also not detected.
Based on the 2008 WHO classification system, the patient was diagnosed with B lymphoblastic leukemia not otherwise specified that was transformed from childhood MDS-refractory cytopenia. She received chemotherapy beginning the day after BM examination. Unfortunately, her general condition deteriorated rapidly due to tumor lysis syndrome during chemotherapy. She expired from hyperkalemia on the third day of chemotherapy.
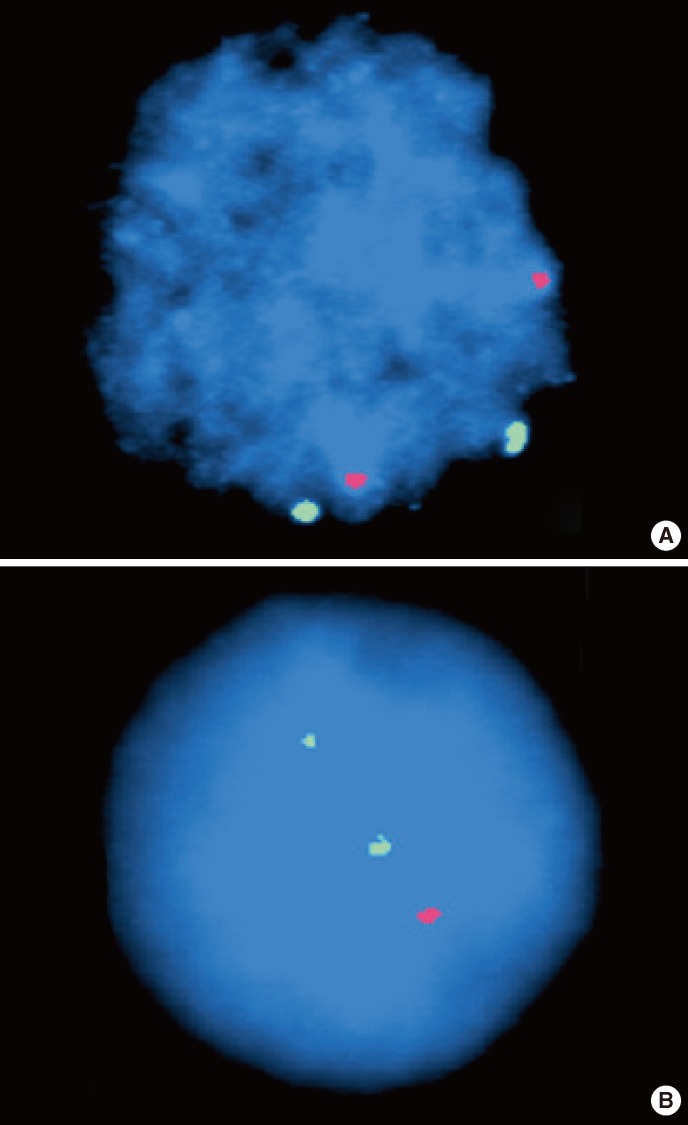
Retrospectively, we analyzed the purified mononuclear cell fractions of the first (MDS-diagnosed) and second (ALL-diagnosed) BM specimens using microarray analysis, Affymetrix Cytogenetics Whole Genome 2.7 Mb Array (Affymetrix, Santa Clara, CA, USA) to identify genetic abnormalities. In the first BM sample, no chromosomal or genetic abnormalities were observed. However, in the second BM sample, we found several large interstitial deletions of 27 Mb and 5.7 Mb on 5q21.2q31.1 and 13q14.1q21.1, respectively, and many microdeletions on chromosomes 5q, 12q, 13q, and 22q. Particularly, a partial homozygous loss of 200 kb was observed in a region of heterozygous loss on chromosome 13q14.1q21.1 in the second BM sample. The chromosome views of the second BM sample using cytogenetic microarray analysis are shown in Fig. 3. To confirm this result, additional FISH analysis of the second BM sample was performed using 2 probes for EGR1 (5q31) and RB1 (13q14), and produced the same result: 5q31.2 was not deleted and 13q14.3 was deleted in 84% of the examined nuclei (Fig. 4).
DISCUSSION
MDS originates from a multipotent hematopoietic stem cell with the potential for myeloid and lymphoid differentiation [11]. Based on flow cytometric immunophenotyping, approximately 85% of blast cells in the leukemic transformation from MDS displayed a myeloid phenotype, while 15% of blast cells displayed a hybrid (myeloid-lymphoid) phenotype. It is extremely rare for only the lymphoid phenotype to be displayed [12, 13]. Previous studies have suggested 2 reasons for the rarity of transformation to ALL from MDS. First, stem cells of MDS may have lymphopoietic potential as well as myelopoietic potential, but there may be specific impairment of lymphopoiesis by intrinsic or extrinsic factors. Second, the rarity of the development of ALL from MDS may reflect a relative deficiency of MDS-derived target cells capable of lymphoid leukemogenesis [14-16].
Transformation to ALL is extremely rare in MDS patients. In adults, several cases of lymphoid transformation from MDS have been reported [17-22]. However, there are only a few case reports of transformation of childhood MDS to ALL worldwide [8-10]. The profiles of our case and the previously reported 3 similar cases are shown in Table 1. In 1996, Bader-Meunier et al. [8] described the clinical, cytological, and cytogenetic features of one childhood MDS patient, a 4-yr-old boy presenting with refractory anemia and excess blasts (RAEB) who had transformed to ALL. The karyotype of the patient was 46 XY, 5q-. In 2007, Goel et al. [9] described a case of 9-yr-old boy with childhood MDS-refractory anemia that transformed into B-precursor ALL after cyclosporine chemotherapy. Cytogenetic analysis of the patient revealed a normal karyotype. In 2010, Gupta et al. [10] reported a 5-yr-old girl who presented with RAEB that evolved into B cell ALL 4 months later. Cytogenetic analysis of the patient was not performed. In contrast to most RCC cases, 2 subsequent cases exhibited erythroid hyperplasia and hypercellularity, similarly to our case [9, 10]. Although most cases of RCC exhibit a normal karyotype irrespective of BM cellularity, the prospective multicenter study EWOG-MDS 98 determined that RCC patients with normal or increased BM cellularity exhibited cytogenetic abnormalities more frequently than in the patients with hypocellular RCC [23].
In our case, conventional karyotyping was first performed to detect cytogenetic abnormalities of RCC and ALL. As there are no cytogenetic abnormalities useful for predicting prognosis, we performed FISH analysis with ALL- and MDS-specific probes. In the retrospective microarray analysis of BM specimens, we discovered cytogenetic abnormalities only in the second BM specimen on chromosomes 5q, 12q, 13q, and 22q.
Recently, the spectrum of genetic abnormalities has shown that MDS can progress to leukemia through the acquisition of multiple genetic abnormalities [24]. Chromosomal abnormalities such as monosomy 7, trisomy 8, and trisomy 21 are common in the transformation of childhood MDS to acute leukemia [25]. In our case, these common abnormalities were not observed. MDS normal karyotype and deletions of chromosome 5q, 20q, and Y are considered favorable prognostic factors, while monosomy 7 and complex abnormalities are considered poor prognostic factors [26]. Particularly, in our case, there was a homozygous loss of 13q14.2, including most of the retinoblastoma gene (RB1). In 50% of de novo MDS cases, chromosomal aberrations and rearrangements involving the RB1 gene in 13q14 are observed [25]. Deletion or translocation involving chromosomal band 13q14, the locus of the RB1 gene, are observed in a variety of hematological malignancies, including AML, plasma cell myeloma, and CLL. Deletion of most of the RB1 gene may be associated with poor prognosis, as the patient died 3 months after the diagnosis of MDS.
We have described a case of an 8-yr-old girl who presented with childhood MDS-RCC that had transformed into ALL. This is the first case report in Korea of transformation into ALL from childhood MDS. Our case shows that there can be similar cases of lymphoid transformation of MDS in the Korean population; it supports, again, the nature of MDS as a pluripotent hematopoietic stem cell disorder. Notably, the patient had a very short progression period compared with the 3 previously reported cases, and multiple cytogenetic aberrations that could not be detected using conventional cytogenetic testing methods. Studies are needed to determine whether cytogenetic analysis methods provide improved means of identifying high-risk patients in childhood MDS.



 XML Download
XML Download