

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
The corpus callosum connects two interhemispheres [1], and agenesis of corpus callosum (ACC) is the most common central nervous system defect [2]. ACC has been reported in 0.3% to 0.7% of the general population and in 2% to 3% of those with developmentally retarded [345]. It may be either complete or partial [6]; the partial form of ACC is also called hypogenesis [7] of corpus callosum. If there are no other anomalies accompanying ACC, then it is defined as isolated ACC.
Since the 1990s, ACC has been diagnosed prenatally through ultrasound screening [8]. Prenatal diagnosis of ACC is considered important because it may be associated with central nervous system abnormalities [910] such as mental retardation, epilepsy, cerebral palsy, and others [1112]. If other anomalies are combined with non-isolated ACC, then outcomes, especially those regarding neurodevelopment, are usually poor [13]. Thus, termination of pregnancy is sometimes recommended when non-isolated ACC is diagnosed [14]; however, this option varies by country.
Long-term studies, especially those regarding neurodevelopmental outcomes, are not available worldwide. Although there is still much controversy [15], most patients with isolated ACC have had good prognoses [16]. In those with isolated ACC, 72.2% showed normal development [16]; in several small sample studies, 100% showed normal development [17]. Normal or mildly delayed neurodevelopment resolves in 67% of isolated ACC, but in non-isolated ACC only 7% resolves [18]. Therefore, distinguishing isolated ACC from non-isolated ACC is clinically important for prenatal counseling.
Few studies have targeted Korean patients regarding the long-term neurodevelopmental outcomes of ACC despite the common practice of targeted ultrasound. Therefore, based on our patients, we aimed to investigate clinical outcomes of prenatally diagnosed ACC and, importantly, to analyze the neurodevelopmental outcomes of those with prenatally diagnosed ACC so that appropriate counseling can be provided.
Materials and methods
We retrospectively reviewed 70 cases of antenatally suspected ACC in fetuses referred to or diagnosed at our center between 2008 and 2015. We excluded 14 patients (20%) with suspected ACC diagnosed at a referral hospital. Thus, only 56 fetuses (80%) with suspected ACC examined at our center were included in this study. Among these, 53 were referred to our center due to abnormal findings on ultrasonography performed at a referral hospital and three were followed-up at our center. The study was approved by the institutional review board.
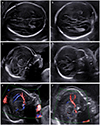
We investigated the following data for each patient: gestational age and ultrasonography findings at the time of referral or first diagnosis of ACC including ventriculomegaly and tear-drop sign; karyotype result if performed; brain ultrasound or magnetic resonance imaging (MRI) findings before or after birth; neonatal outcome; postnatal diagnosis; delivery mode; delivery indication; gestation age at delivery; neonatal birth weight; and neonatal intensive care unit admission. Gestation age at diagnosis was recorded based on the gestation age at the time of transfer or time of diagnosis in our hospital. The diagnosis of ACC was made by using indirect signs of ACC on ultrasound, including the presence of a tear-drop sign and absence of cavum septum pellucidum (CSP), or by direct nonvisualization of the corpus callosum on the sagittal plane (Fig. 1). Ventriculomegaly, an enlargement of the posterior horn more than 10 mm, was not regarded as an associated anomaly because it may be one of the characteristics of ACC.
Isolated ACC was defined when there was no other anomaly on ultrasound or MRI based on prenatal or postnatal findings, respectively. The partial or complete form of ACC was not separately distinguished because they showed no significant difference with respect to functional subtyping of the callosum and neurological outcome [19].
We analyzed the neurodevelopmental outcomes based on medical records assessed and described by the pediatric neurology or rehabilitation doctors if delivery was performed in our center. Neurodevelopmental outcomes were followed-up until approximately age 3 years. To supplement neurodevelopmental outcomes of isolated ACC, we also tried analyzing neurodevelopmental outcome of postnatally diagnosed ACC patients (n=9) who primarily visited the pediatric neurology or rehabilitation department of our center. We classified neurodevelopmental outcomes into four categories; normal, mild, moderate, severe. ‘Mild’ included children with neurodevelopment delay within six months, ‘Moderate’ with 6 months to 12 months delay, and ‘Severe’ with neurodevelopment delay of more than 12 months. The assessment of neurologic development was performed through the clinical history taking of development milestones. Objective development evaluation includes hand evaluation, Alberta infant motor score, Activities of Daily Living evaluation, Denver Developmental Screening Test, etc. but which were not performed on every child. Follow-up evaluation for all cases with prenatally diagnosed ACC (and postnatally diagnosed ACC) was performed by pediatricians and pediatric neurologists. The final neurodevelopmental outcome was assessed by two pediatric neurologists (JWL and JHL) by review of medical record.
Since we could not obtain results by directly calling the patient, which was not permitted by IRB, we endeavored to ascertain follow-up or delivery results by calling the referral institution if delivery was not performed in our institution. “Follow-up loss” meant that patients did not go back to the original clinic or give birth there. Patients of hospitals that no longer exist or of hospitals that could not provide information were classified as “unknown.” To check whether ACC was present, brain ultrasound was performed after birth. Some patients underwent brain MRI when other brain anomalies were suspected.
Results
This study included 56 patients with ACC prenatally diagnosed by ultrasound examination. Through prenatal diagnosis, we classified 56 patients as having either isolated or non-isolated ACC and summarized the clinical characteristics (Table 1). Among these 56 patients, 29 (51.8%) were considered to have isolated ACC and 27 (48.2%) were considered to have non-isolated ACC. These classifications were based on prenatal sonographic findings alone and did not consider the postnatal diagnosis. Ventriculomegaly and a tear-drop sign were observed in approximately 70% to 80% cases of prenatally diagnosed ACC. The median atrial diameters were 16.3 mm and 12.6 mm for isolated ACC and non-isolated ACC, respectively. Prenatal chromosomal analyses were performed for 6/29 (20.7%) in the isolated ACC group and for 8/27 (29.6%) in the non-isolated ACC group. With the exception of unknown results, all fetuses in the isolated ACC group showed normal chromosomes and one of eight (12.5%) in the non-isolated ACC group had a chromosomal abnormality (trisomy 18). According to follow-up results, the rates of referral back to the hospital were 17.2% for the isolated ACC group and 7.4% for the non-isolated ACC group.
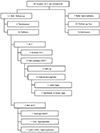
We investigated the follow-up results of these fetuses with prenatally diagnosed ACC (Fig. 2). Twelve deliveries were performed in our center, including two terminations that occurred during the early study period; the other 44 patients were referred back to the original clinic or lost to follow-up. Among 44 patients who did not undergo delivery at our hospital, seven were referred back to the original clinic due to their long distance from our center and were confirmed as having delivered the pregnancy. Twenty-three patients did not go back to the original clinic or did not give birth there and were considered lost to follow-up. This group was suspected of undergoing termination. The remaining 14 patients of no longer existing hospitals or hospitals that could not provide information were classified as unknown. These follow-up data indicate that the overall percentage of follow-up loss for prenatally diagnosed ACC was 41%, but it increased up to 66% when the unknown category was included.
Clinical characteristics and outcomes for 12 fetuses with ACC delivered at our center are presented in Table 2. Among 10 live fetuses delivered, four had isolated ACC, three had non-isolated ACC, and the rest were found to have outcomes unrelated to ACC after birth. Excluding termination cases, three out of four suspected isolated ACC cases during the prenatal period were actually confirmed as isolated ACC postnatally, but the other one was finally diagnosed as non-isolated ACC combined with semilobar to lobar holoprosencephaly. On the contrary, one out of the six cases considered to be non-isolated ACC was finally proven to be isolated ACC because the degree of enlarged mega cisterna magna was found to be normal on postnatal ultrasound. In the non-isolated ACC group, two patients had holoprosencephaly (patients 2 and 7) and one had cleft lip and right inguinal hernia (patient 10). One of two patients had lobar holoprosencephaly, was followed-up for delayed development, and was lost to follow-up after 17 months (patient 2); another had semilobar to lobar holoprosencephaly and is currently being observed for symptomatic localization-related epilepsy (patient 7). Among our study population, there were two patients who were initially suspected with ACC but were subsequently diagnosed as having complications of fetal hydrops and coarctation of the aorta. ACC was originally diagnosed because the CSP was not visible on ultrasound.
Among the four diagnosed with isolated ACC, one was transferred to another hospital because of the mother's personal preference (patient 3). The other three (patients 1, 4, and 6) were followed-up for at least 12 months and showed no developmental delay, suggesting that at least 75% of patients with isolated ACC had normal neurodevelopment.
Table 3 shows the summary of clinical characteristics of 10 live births (with the exception of two terminations). Median gestation age at diagnosis was 27 weeks. However, with the exclusion of four patients admitted for delivery near term or at term, the median gestation age at prenatal diagnosis was 22 weeks. Chromosomal study results were available for 8 of the 10 patients who delivered. Two did not undergo a chromosomal study (patients 1 and 3) because they planned for delivery regardless of abnormal chromosome results of the fetuses. Of these eight who underwent chromosomal study, a normal karyotype was found in seven fetuses and one had chromosomal anomalies: arr Xp11.22(53,166,281–53,427,895)x1 (patient 7). One patient who had a chromosomal abnormality had non-isolated ACC and semilobar to lobar holoprosencephaly and was small for gestational age; this patient later presented with epilepsy.
Supplementary analysis including postnatally diagnosed isolated ACC, in which follow-up was performed at least 9 months to maximum 4 years in the pediatric neurology or rehabilitation department, demonstrated that 5 (55.6%) showed normal developmental outcome, 2 (22.2%) showed mild developmental delay, another 2 (22.2%) showed moderate developmental delay, and none showed severe developmental delay.
Discussion
Our data demonstrated that approximately 75% of isolated ACC showed normal neurodevelopment. Although limited in number, our finding is in line with previous studies reporting that the rate of normal neurodevelopment for 16 patients with isolated ACC was 71.2% [20]. In our study, we also found that the follow-up loss rate for prenatally diagnosed ACC is substantial even for isolated ACC, despite our endeavor to counsel these patients. In fact, each physician in our center generally provides information about normal neurodevelopmental outcomes being more than 80% for isolated ACC based on previous works [1721]. Therefore, a conundrum beyond proper counseling at the time of prenatal diagnosis of ACC and subsequent follow-up remains in our country. However, the impact of proper counseling for prenatally diagnosed ACC was demonstrated by a study performed in another country. For instance, in France, the termination rate for ACC has decreased by almost 17%, from 13/35 (37.1%) in 2000–2003 to 9/44 (20.5%) in 2003–2006 [22]. This decrease seems to be attributable to proper counseling based on information regarding the good prognosis for isolated ACC. Therefore, although it is difficult to lower the termination rate for non-isolated ACC with associated anomalies, appropriate counseling regarding the higher percentage of normal neurodevelopment for isolated ACC is expected to reduce that termination rate.
When checking brain structures during the mid-trimester fetal anatomy survey, there is a tendency to obtain a mainly transverse view. However, a mid-sagittal view is more accurate for determining ACC. Moreover, indirect signs of ACC are either absent or not clearly visible at the time of mid-trimester screening ultrasonography, which is performed before 24 weeks of gestation in many cases [23]. The gestational age at diagnosis of ACC in other countries is 23 to 26 weeks [2425], which appears rather late compared to that in our country, although this is probably due to the more frequent use of ultrasound in our country. ACC is frequently associated with ventriculomegaly; our data show that up to 70% to 80% of those with ACC have ventriculomegaly. Therefore, fetuses with ventriculomegaly shown by prenatal ultrasound require a more detailed examination for the existence of the corpus callosum. In fact, it is reported that there is a callosal abnormality in 13% of ventriculomegaly cases [18]. When indirect signs of ACC are suspected on mid-trimester ultrasound screening, short-term follow-up and repeat ultrasound with a mid-sagittal view are recommended.
Sometimes it may be difficult to confirm ACC as being isolated on prenatal ultrasonography alone. Our data also showed that one out of four cases of prenatally diagnosed ACC was subsequently found to be non-isolated after birth. In reality, it was reported that approximately 5% to 20% of cases are misdiagnosed as isolated ACC during the prenatal period [26]. On the contrary, 16% (1/6) of non-isolated ACC cases were finally proven to be isolated ACC, indicating that false-positive diagnoses are possible. To overcome this kind of false-negative or positive diagnosis, differential diagnoses such as septo-optic dysplasia and holoprosencephaly should be considered if there is no CSP on ultrasonography; in addition, some ancillary tools such as 3D ultrasonography or fetal MRI have been suggested. A study by Pashaj et al showed that 3D ultrasonography is helpful in the diagnosis of ACC [27]. Recently, several studies demonstrated the usefulness of fetal MRI to improve the sensitivity of the ACC diagnosis and to affirm associated anomalies [2829]. Therefore, fetal MRI is recommended when necessary. In our study population, fetal MRI was performed for only 16% during the perinatal period. This low rate of fetal MRI performed in our center may reflect the specific laws of Korea, which prohibit abortion solely for fetal indications.
One-third of ACC cases are isolated ACC and the other two-thirds are non-isolated ACC [25]. In our study of 56 patients, 51.8% were considered to have isolated ACC and 48.2% were considered to have non-isolated ACC prenatally. Postnatally, 57% (4/7 cases) in this study had isolated ACC. However, this result might have been affected by selection bias, because these cases were found in patients who had been prenatally suspected to have isolated ACC and were followed-up after delivery.
The limitations of our study include the high rate of follow-up loss and insufficiency of long-term follow-up. There were 37 fetuses with unknown results. Data regarding the results for these patients could not be obtained for 14; the remaining 23 did not return to the original centers from which they were referred, and they were confirmed to be follow-up losses. There is a possibility that these patients underwent delivery in referred hospitals or other locations other than our center, but there is a high possibility that the majority chose termination. Therefore, adequate counseling regarding ACC is necessary to reduce the termination rate. Also, long-term follow-up at least until the age of beginning school [30] is necessary because there is a possibility that some neurodevelopmental delay may develop with time; the current follow-up duration is too short. In addition, large numbers of children with isolated ACC are needed for future studies. The incidence of ACC is less than 1%; therefore, multicenter studies of ACC and regular follow-ups conducted through consultation with pediatrics, neurology, or rehabilitation medicine departments are essential. Future studies will be meaningful for determining the prognosis of ACC and will be helpful for counseling patients.
It is noteworthy that we also supplemented the neurodevelopmental outcome of postnatal isolated ACC to partially overcome the limitation of small number in our study population. Considering that outcome of postnatally diagnosed ACCs could be comparatively unfavorable to prenatally diagnosed ACCs as they visit the hospital due to the presentation of symptoms, the findings that 55.6% and 75% of isolated ACC (postnatally and prenatally, respectively) showed normal neurodevelopmental outcome may be in accordance each other.
In summary, we demonstrated that neurodevelopmental outcomes for those with isolated ACC are favorable. However, we found very few studies on ACC in our country. A recent article from the perspective of rehabilitation investigated the neurodevelopmental outcomes of corpus callosal anomaly in Korea, but this study also included hypoplasia of corpus callosum as well as ACC [31]. Although limited in number, our single-center study has modest implications regarding neurodevelopmental outcomes of isolated ACC in Korea. We believe this clinical information is helpful for properly counseling these patients.



 XML Download
XML Download