

 PDF
PDF ePub
ePub Citation
Citation Print
Print



PART I
Introduction
Autophagy is an evolutionarily conserved catalytic process for maintaining cellular homeostasis by which cytoplasmic components including damaged macromolecules and organelles are degraded, thereby providing the new building blocks for cellular recycling. The role of autophagy includes adaptive responses to nutrition deprivation and quality control of intracellular proteins and organelles. Autophagy is also related to cellular defense mechanisms against microorganism invasion of the host and thus plays an important role in innate and acquired immunity. As previous studies considered autophagy as another type of cell death mechanism, it was called type II cell death [1]. However, accumulating evidence has suggested that autophagy usually exerts a prosurvival function in response to stress signals from the intracellular or extracellular microenvironment, and thus the classification of autophagy as autophagic cell death has now been challenged [2]. According to the intracellular organelles or substrate for autophagic degradation, autophagy is termed mitophagy (mitochondria), aggrephagy (protein aggregates), xenophagy (intracellular pathogen), or lipophagy (lipids). Recently, it was also found that the recognition of autophagy substrate is conducted in a selective manner by cargo-specific factors [3].
Autophagosomes were first observed by electron microscopy in mammalian cells in the 1962 [4], and in 1963, the term autophagy was coined by de Duve, who discovered two cell organelles, peroxisomes and lysosomes [5]. However, it was not until the 1990s that autophagy-related genes in yeast were identified and the morphologic change of autophagy in yeast was demonstrated [6]. Until now, approximately 35 different proteins have been known to be involved in this orchestrated process in yeast, and at least 11 have orthologs in mammals [7]. Beclin 1 and microtubule-associated protein 1 light chain 3 (LC3), among the molecules most studied in autophagy in mammals, are orthologs of Atg6 and Atg8 in mammals, respectively. In the last decade, there was a substantial expansion of knowledge regarding the molecular mechanisms of autophagy pathways and the physiological role of autophagy among eukaryotes. Defects in autophagy-related genes in humans are now known to be directly related to the pathogenesis of certain diseases such as breast carcinoma and Crohn's disease [89]. Furthermore, aberrant autophagic activity has also been implicated in various diseases including neurodegenerative diseases, infectious or inflammatory diseases, malignancies, and autoimmune diseases [10]. For example, Alzheimer's disease is associated with increased accumulation of autophagosomes in human brain tissue resulting from impaired autophagic activity [11]. And, several important bacteria (e.g., Mycobacterium tuberculosis and Listeria monocytogenes) and viral (herpes simplex virus-1) pathogens are degraded by xenophagy, showing a protective role of autophagy against invasion of microorganisms [12]. Without exception, autophagy is also involved the events of reproduction and pregnancy. It was already known that fertilization of oocytes was accompanied by autophagy induction, and thus autophagy is critical for preimplantation mouse embryo development [13]. Mice models of defective autophagy genes also exhibit neonatal lethality [14].
However, we recognize that our understanding of autophagy in the placenta is just at the beginning. Considering the importance of the placenta in maintaining a successful pregnancy, autophagy research on the placenta might provide clues to solve the many unanswered questions about pregnancy complications related to abnormal placental development. This part I covers general information on autophagy and next part II will provide summaries from articles on autophagy in human placenta. Finally, we tried to provide useful information to researchers pursuing autophagy research in relation to human pregnancy and its complications.
Autophagy overview
1. Steps and machinery of autophagy
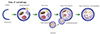
The steps of autophagy begin with the formation of an isolation membrane (phagophore), which elongates into an autophagosome with a double membrane. The mature autophagosome engulfs cytosolic cargo such as damaged macromolecules and organelles and, after fusion with lysosome, forms an auto phagolysosome, and finally the autophagosome cargo is digested by a lysosomal protease (Fig. 1). There are large numbers of stimuli that induce autophagy. Starvation or nutritional stress is the most well-known autophagy inducer. In addition, endoplasmic reticulum (ER) stress, immune signals, mitochondrial damage, hypoxia, and redox stress are well-known inducers of autophagy in various cells [15]. Accordingly, numerous upstream signaling pathways including phosphatidylinositol 3-kinase (PI3K)/Akt, AMP-dependent protein kinase (AMPK), growth factor signaling, mitogen-activated protein kinases, inositol triphosphates, and calcium signaling are involved in the process of autophagy [15]. The whole picture of this complex molecular network is well delineated in several review articles [16] and briefly summarized in the following section.
The most renowned and crucial upstream molecular pathway is that of mTOR, which resides in the macromolecular mTOR complex 1 (mTORC1) and blocks autophagy induction in nutrition-replete conditions for physiologic homeostasis. However, in a state of nutrition deprivation, mTOR activity is suppressed, which leads to the activation of UNC-51–like kinase 1 (ULK1), an initiating step of the autophagic process. Autophagy is also regulated by beclin 1 interacting complex, consisting of beclin 1, the Bcl-2 family, the class III PI3K (vesicular protein 34, VPS34), and ATG14L [17]. Stimulation of this complex increases production of phosphatidylinositol-3-phosphate, which regulates the formation of autophagosomes. Meanwhile, the PI3K/Akt signaling pathway negatively regulates the beclin 1 complex and stimulates mTOR, thereby inhibiting autophagy. Autophagosome elongation occurs via two ubiquitin-like conjugation systems, the Atg5-Atg12 conjugation system and the LC3 (the mammalian homolog of yeast Atg8) conjugation system [18]. LC3-II, which is formed by conjugation of phosphatidylethanolamine from LC3-I, is incorporated into autophagosomes and thus recognized as a hallmark of autophagosome formation.
2. Regulation of autophagy and its signaling pathway
As stated above, autophagy can be induced by diverse stimuli including nutritional deprivation, ER stress, infection and inflammation, hypoxia, redox stress, and mitochondrial damage. Accordingly, various signaling pathways related to each stimulus are involved in this process of autophagy induction.
1) Nutritional stress
Nutritional deprivation is one of the most well-known autophagy inducers, and mTOR inhibition is its best characterized mechanism. In the presence of growth factors such as insulin or insulin-like growth factors, stimulated Akt and extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) can phosphorylate and disrupt the tuberous sclerosis complex 1/2 (TSC1/TSC2), which activates mTOR inhibition and thus inhibiting autophagy [19]. However, when growth factors are depleted, the active TSC1/TSC2 complex suppresses mTOR activity, followed by induction of autophagy. The second most critical molecule that regulates autophagy in response to metabolic stress is via the AMPK pathway. AMPK checks the energy status by sensing the AMP:ATP ratio of the cell. AMPK is also activated by several intracellular stresses including high intracellular calcium levels as well as energy depletion via diverse upstream kinase activation [20]. For example, energy depletion activates liver kinase B1 and high cytosolic Ca++ induces calcium/calmodulin kinase kinase-β. This activated kinase stimulates AMPK, which induces autophagy via mTORC1 inhibition (via phosphorylation of TSC2 and the regulatory-associated protein of mTOR, Raptor).
2) ER stress
Autophagy is also induced by the unfolded protein response, an important ER stress pathway, and several ER-associated proteins are involved in its signaling pathway. The major ER proteins that regulate autophagy include protein kinase R-like eIF2α kinase (PERK), activating transcription factor-6 (ATF6) and IRE1 (inositol-requiring enzyme 1). PERK induces transcriptional activation of LC3 and Atg5 in response to hypoxia [21]. It was also demonstrated that ATF4 induced by PERK via eIF2α phosphorylation is associated with increased LC3 expression [22]. IRE1, a serine/threonine kinase, can stimulate autophagy via activation of Jun N-terminal kinase 1 (JNK1) or negatively regulate autophagy by action of X-box binding protein 1 [15].
3) Immune signal or inflammatory stimuli
Autophagy induction during infection is mediated by cytokines such as interferon-γ as well as pathogen recognition receptors. Indeed, Toll-like receptors trigger autophagy during innate and adaptive immune responses. For example, lipopolysaccharide induces autophagy via Toll-like receptor-4 in primary human macrophages [23]. Whereas Th1 cytokines such as interferon-γ and tumor necrosis factor (TNF)-α induce autophagy, Th2 cytokines including interleukin (IL)-4 and IL-10 inhibit autophagy [24]. The signaling pathway underlying how inflammatory signals trigger autophagy seems to differ in different cells. For example, in human vascular smooth muscle cells, TNF-α upregulates autophagy via the JNK pathway and inhibition of Akt [25], and in the MCF-7 human breast cancer cell line, the ERK1/2 pathway is involved in TNF-α–induced autophagy [26]. Autophagy also regulates the secretion of numerous cytokines including IL-1, IL-18, and TNF-α [24].
4) Hypoxia and anoxia
There are several mechanisms by which hypoxia or anoxia induce autophagy. In general, for moderate hypoxia (1% to 3% oxygen), the hypoxia-inducible factor 1-α (HIF1-α) pathway is predominantly used. In brief, stabilization of HIF1-α by hypoxia activates the transcription of Bcl-2/adenovirus E1B 19 kDa-interacting protein (BNIP)3/BNIP3L, which weakens interaction between beclin 1 and Bcl-2 and subsequently elicits autophagy [27]. Furthermore, it was also demonstrated that increased Redd1 by HIF1-α activates TSC1/2 resulting in the inhibition of mTORC1 activity followed by increased autophagy [28]. Severe hypoxic conditions or anoxia (<0.1% oxygen) can induce a HIF1-α–independent autophagic response through activation of AMPK, which inhibits mTOR activity [15] or through the unfolded protein response pathway [29]. In addition, hypoxia enhances the transcription of LC3 and ATG5, which are crucial autophagy genes [21].
5) Redox stress
Multiple mechanisms are involved in redox stress-induced autophagy. Increased reactive oxygen species levels can induce the activation of PERK and JNK1, which mediate autophagy induction [30]. In addition, exogenous hydrogen peroxide can directly activate Atg4 protease and thus increase the production of mature LC3 [31]. Direct or indirect inhibition of mTOR by oxidative stress was also demonstrated [15].
3. Assessment of autophagy in mammalian cells
1) Electron microscopy
Conventional electron microscopy is obviously the most standard and reliable method to assess autophagy. Because it is difficult to differentiate by transmission electron microscopy (TEM) between autophagosomes and autophagolysosomes, which are double- and single-membrane structures, respectively, containing cytoplasmic organelles, the term, autophagic vacuoles is commonly used to indicate these structures together. The quantification of autophagy by TEM can be achieved as the ratio of the area of autophagic vacuoles among the total cytoplasmic area [34]. However, TEM has a limitation in that it requires highly skilled experts and is time-consuming. Furthermore, sometimes other organelles such as the ER can be mistaken for autophagic vacuoles by TEM, making it not objectively quantitative.
2) LC3-positive puncta
LC3-positive puncta, a marker of the induction of autophagy, can be observed by immunofluorescence using anti-LC3 antibodies. LC3, fused with green fluorescent protein (GFP) is commonly for in vitro assessment of autophagy. The real-time observation of GFP-LC3 localization is also possible. It was demonstrated that overexpression of GFP-LC3 itself does not modulate endogenous autophagy activity. GFP-LC3 transgenic mice can be used for in vivo analysis of autophagy [35]. Recently, autophagic flux ex vivo in retinal explants from GFP-LC3 transgenic mice using lysosomal inhibitors was also demonstrated [36].
3) Tandem fluorescent tagged LC3 method
A novel tandem fluorescent tagged LC3 method was proposed to avoid a pitfall in the interpretation of an increase in the number of LC3-positive puncta, which might resulted from the accumulation of autophagosomes rather than the formation of autophagosomes. The limitation of GFP itself for the degradation of lysosomal protease could also be overcome using the tandem fluorescent tagged LC3 vector [37]. In this vector, red fluorescent protein, which is resistant to lysosomal degradation, is also fused with GFP-LC3. According to the stage of autophagy, autophagosomes appear in yellow puncta, whereas mature autophagolysosomes appear red because of the quenching of GFP in the acidic milieu of the lysosome [38].
4) Western blot for LC3
LC3, a mammalian ortholog of yeast Atg8, is the most frequently used to assess autophagy in cells. Because autophagy is a dynamic process, it is important to understand the basis of the expression of LC3 and the principles of interpretation. In fact, LC3 can localize to any types of autophagic membrane including the phagophore, autophagosome, and autolysosome during the autophagic process [3940]. Pro-LC3 is cleaved to LC3-I by an Atg4 family protease immediately after synthesis [39]. During autophagy, LC3-I is converted to LC3-II with conjugation to phosphatidylethanolamine by the action of Atg3 and the Atg12-Atg5-Atg16L1 complex, and thus LC3-II is localized in the autophagosomal membranes. Finally, LC3-II is also degraded by autophagy in autophagolysosomes. The amount of LC3-II or LC3 conversion (LC3-I to LC3-II) is often used to monitor autophagy, indicating autophagosome formation. However, it should be noted that increased LC3-II levels can be associated with either increased autophagosome formation or reduced turnover [37]. Moreover, because autophagy is a dynamic process regulated by both the on and off rate, the amount of LC3-II does not necessarily reflect the degree of autophagic activity at a certain time point [4142]. Thus, autophagic flux, defined as the dynamic process of autophagy, is usually recommended to prove autophagosome formation by comparing the amount of LC3-II in the presence or absence of a lysosomal inhibitor such as bafilomycin A1 (a vacuolar H+-ATPase inhibitor) or pepstatin A (lysosomal protein inhibitor). A detailed interpretation of this autophagic flux assay has been well described [43]. Of note, there are some technical issues, which can provide several troubleshooting tips in western blotting for LC3. Although the actual molecular weight of LC3-II is greater than that of LC3-I owing to its binding to phosphatidylethanolamine, the extreme hydrophobicity of LC3-II means it moves faster than LC3-I on sodium dodecyl sulfate polyacrylamide gel electrophoresis. A second point is that certain anti-LC3 antibodies are less sensitive for detecting LC3-I, especially with the semi-dry transfer method [44]. Additionally, it was also reported that the use of a polyvinylidene difluoride membrane rather than a nitrocellulose membrane was associated with successful western blotting for LC3 [37].
5) p62
Another widely used marker of autophagy is p62, also called as sequestosome 1, which is also an adaptor molecule implicated in the targeting of cargo for autophagosomes. That is, p62 interacts with polyubiquitinated protein aggregates, thereby facilitating these aggregates for degradation at the autolysosome through direct binding with LC3 by a short LC3 interaction region [45]. Because p62 itself is degraded by autophagy, p62 accumulation represents defects in selective autophagy of ubiquitinated aggregates [43]. However, measurement of p62 has some pitfalls. Since p62 is also degraded by ubiquitin-proteasome system as well as autophagy, its level may be increased when the proteasome is inhibited. In addition, the transcription of p62 itself could be increased by oxidative stress. Therefore, an additional method to monitor autophagy is generally needed [46].
4. Cross-talk between autophagy and apoptosis
Accumulating evidence has shown that there are multiple interactions between autophagy and apoptosis. These interactions may be synergistic or antagonistic in the context of cell type and stimuli [147]. Several scenarios have been proposed so far.
First, autophagy often precedes apoptosis in most cells [47]. In this scenario, it seems that autophagy plays a cytoprotective role by eliminating the proapoptotic stimuli such as damaged mitochondria. Therefore, autophagy sets the threshold during apoptosis for cells to escape from death signals and to maintain their survival in stress conditions. At this point, the level of stress is considered not yet lethal. For example, rapamycin (pro-autophagic) pretreatment can decrease the mitochondrial load by upto 50% while reducing the susceptibility of cells to mitochondrial outer membrane permeabilization-dependent apoptotic signals [47]. In fact, this protective effect was mediated by enhanced mitophagy, which reduced cytochrome c release and caspase activation [48]. Similar protective effects of enhanced autophagy were also demonstrated in palmitate-induced apoptosis in hepatocytes or oxidative stress-induced apoptosis in a rat model of Parkinson's disease [4950]. However, when excessive toxic signals threaten cells beyond this defense, ultimately apoptosis as well as autophagic cell death result. In this context, the same stimuli can induce either apoptosis or autophagy and many signaling pathways such as p53, Bcl‐2 homologous 3-only proteins, death-associated protein kinase, and JNK are involved. A typical example is p53, which upregulates pro-autophagic transcription including AMPK and damage-regulated autophagy modulator 1 (DRAM-1) as well as induction of apoptosis by transcriptional activation of multiple proapoptotic genes upon cellular stressors such as DNA damage and ischemia-reperfusion [47].
Second, autophagy and apoptosis may antagonize each other. In this inhibitory cross-talk, autophagy reduces the propensity of cells to elicit apoptosis. During intrinsic apoptosis when mitochondrial outer membrane permeabilization releases catalytic hydrolase and caspase activators, selective autophagy of mitochondrial (mitophagy) can modify apoptosis by increasing the threshold for apoptosis [51]. In this context, inhibition of autophagy can increase apoptosis. For example, it was reported that specific inhibition of autophagy by knockout of Atg7 in mice hepatocytes increased TNF-dependent liver injury via increased caspase-8 activation [52]. On the contrary, activation of apoptosis could inhibit the autophagy machinery of cells. Caspase activation during apoptosis can digest several proteins that are essential for the autophagic machinery to operate. For example, it was reported that apoptosis induced by the proapoptotic protein Bax suppressed autophagosome synthesis by caspase-mediated cleavage of beclin 1, which is the initial step of autophagy activation [53].
Third, induction of autophagy can facilitate the activation of apoptosis. In this scenario, autophagy develops as a primary response to stimuli and then triggers apoptosis. For example, in CD4+ T cells, autophagy induced by human immunodeficiency virus-1 envelope glycoproteins was required for apoptosis based on the observation that pharmacologic inhibition of autophagy with 3-methyladenine or small interfering RNAs (siRNAs) for beclin 1 or Atg7 genes totally inhibited the apoptotic process [54]. Similarly, it was also demonstrated that inhibition of autophagosome formation by depletion of Atg5 or Atg3 results in a marked suppression of caspase-8 activation and apoptosis in mouse embryonic fibroblasts cell lines [55]. It was also suggested that autophagy may stimulate apoptosis by the depletion of endogenous inhibitors of apoptotic pathways [51].
In fact, the proteins essentially involved in autophagy are known to have proapoptotic actions. Therefore, it is now considered that some ATG genes are not exclusively involved in autophagy but also associated with other cellular functions such as endocytosis as well as apoptosis [56]. Recently, multiple checkpoints in which autophagy and apoptosis are intertwined have been demonstrated at the molecular level as detailed below.
1) Beclin–Bcl-2 interaction
Beclin 1 performs a critical role in autophagosome formation by interaction with VPS34. When nutrients are sufficient, this pro-autophagic action of beclin 1 is inhibited by direct binding with Bcl-2, a well-known antiapoptotic protein. During stress conditions, beclin 1 is released from Bcl-2 interaction and induces autophagy. Meanwhile, Bcl-2 inhibits apoptosis by blocking Bax activation and thus preventing mitochondrial outer membrane permeabilization and the subsequent release of cytochrome c. So far, several molecular mechanisms have been reported to be involved in the dissociation of beclin 1 and Bcl-2 binding [5157]. For example, death-associated protein kinase phosphorylates beclin 1 and interferes with its interaction with Bcl-2, allowing induction of autophagy.
2) The tumor suppressor protein p53
p53 also regulates autophagy in a stimulatory or inhibitory manner. Such dual actions of p53 on autophagy are dependent on the cellular location of p53, which is usually present in the cytosol where it inhibits autophagy as well as apoptosis by PI3K/Akt activation. However, in conditions of cellular stress, p53 moves into the nucleus and transactivates several genes involved in autophagy, such as death-associated protein kinase [58], resulting in enhanced autophagy. It also induces autophagy by increasing phosphorylation of Bcl-2, which weakens its interaction with beclin 1.
3) P62 (sequestosome 1)
P62, a multifunctional protein that binds to ATG/LC3 and exerts proteasomal activity and autophagy, is also interconnected with apoptosis through activation of the caspase 8 complex [59]. Flice inhibitory protein, a known inhibitor of apoptosis, also inhibits autophagy by blocking ATG3 conjugation to LC3 [60]. In addition, it was also suggested that certain Atg proteins may play dual roles in autophagy and apoptosis. For example, Atg5, which regulates autophagy, can be subject to calpain-dependent cleavage to generate a proapoptotic truncation product (tAtg5). This cleavage product promotes apoptosis by binding to and inhibiting antiapoptotic proteins such as Bcl-XL [61]. Likewise, accumulating evidence has shown that several proteins known to regulate autophagy are involved in apoptosis and vice versa [57].
5. Autophagy and human disease
A growing number of researches have suggested that autophagy is implicated in many human diseases including neurodegenerative disease, metabolic disease, cancer, and inflammation. Recently, it was also known that autophagy is involved in physiologic aging process of humans. Here, a brief summary of current evidence on of the role of autophagy in human disease is provided.
1) Neurodegenerative disease
As neurodegenerative disease is commonly characterized by mitochondrial dysfunction and the accumulation of protein aggregates, autophagy is considered to be a protective mechanism of the pathophysiology of such neurodegenerative disease.
It is well known that Alzheimer's disease is associated with increased accumulation of autophagosomes in human brain tissue resulting from impaired autophagic activity [10]. Direct causal relationship between defective gene function and autophagy was also shown in recessive familial form of human Parkinson's disease, in which the mutation of PTEN-induced putative kinase protein 1 (PINK1) and PARK2 (the genes encodes parkin) proteins are involved in the mobilization of dysfunctional mitochondria to autophagosome, was demonstrated [62]. Animal experiments using mice also suggest that defective autophagy genes promote age-dependent neurodegeneration and pharmacologic treatment with autophagy enhancer can attenuate symptoms from neurodegeneration [63].
Autophagy itself is impaired by the protein aggregates from several neurodegenerative diseases. For example, Alzheimer' disease is associated with excess hyperphosphorylated microtubule associated protein tau. This leads to formation of neurofibrillary tangles and the accumulation of beta amyloid peptide, which impairs lysosomal function and autophagic clearance [10]. Similarly, Parkinson's disease is characterized by the accumulation of α-synuclein aggregate in Lewy bodies—neural cytoplasmic inclusions that impair autophagic clearance [64].
2) Infection and inflammation
The role of autophagy in infection or inflammation can be categorized into several aspects. First, autophagy can directly degrade intracellular microorganisms through a process called xenophagy. Such process can involve autophagosomes that engulf certain types of microorganisms, which include shigella flexneri, salmonella enteric, group A streptococcus, mycobacterium tuberculosis, listeria monocytogenes, herpes simplex virus, toxoplasma gondii, etc. [10]. Other than that, it can enhance phagosome fusion with lysosome, thereby killing the organisms in toll-like receptor signaling pathway [65].
Autophagy also plays an important role in adaptive immunity by enhancing antigen presentation and lymphocyte development. For example, it was shown that deletion of Atg5, a key autophagy gene, in dendritic cells manifested impaired CD4+ T cell priming after herpes simplex virus infection [66]. Similarly, macrophage-specific Atg5 deletion was found to be associated with increased susceptibility to mycobacterium tuberculosis infection [67]. In this context, pharmacologic upregulation of autophagy is considered as an important strategy in immune response to the invasion of microorganisms and furthermore, invoked to the field of vaccine development. Indeed, it was demonstrated that rapamycin-induced autophagy enhanced mycobacterial antigen Ag85B presentation and promotes the efficacy of BCG vaccine in mouse dendritic cells [68]. Autophagy can suppress immune response by downregulation of proinflammatory cytokine response to invading pathogen.
Recently, association between derangement in autophagy genes and susceptibility to certain infections or inflammatory diseases are also identified and among them, mutations in autophagy regulators and Crohn's disease are best characterized. Single nucleotide polymorphisms in ATG16L1, nucleotide-binding oligomerization domain containing protein 2, and immunity-related p47 guanosine triphosphate M protein were identified to increase the risks Crohn's disease by genomewide association studies [69].
3) Malignancy
In contrast to the most beneficial effect of autophagy in neurodegenerative disease and infection or inflammation conditions, the role of autophagy in malignancy is more complex and can interact as a double-edged sword and multifactorial influences depending on tumor type or context. Earlier study by Liang et al. [9] showed that beclin1 was mono-allelically deleted in 40% to 75% of human breast and ovarian cancers and increased beclin 1 activity inhibited MCF7 cellular proliferation, indicating decreased expression of autophagy proteins may contribute to the pathogenesis of breast and other human malignancies. Mice experiments also proved that monoallelic loss of beclin1 lead to tumoriogenesis [70]. Given the fact that defective autophagy induces mitochondrial dysfunction, making normal cells subject to DNA damage and instability, autophagy may accomplish its role as a safeguard to prevent the occurrence of malignancy.
However, from the view point of cancer cells already established, autophagy can be used as survival strategies to enhance tumor proliferation and invasion and to acquire resistance against chemotherapy or radiation. For example, in primary pancreatic cancer and its cell lines, which showed elevated autophagy under basal conditions, autophagy inhibition by silencing essential autophagy gene ATG5 or pharmacologic agent resulted in significant growth suppression [71]. Of note, autophagy confers the anticancer drug resistance and therefore inhibition of autophagy may sensitize the tumor cells to anticancer therapy. Indeed, several autophagy inhibitors, such as 3-methyladenine, chloroquine and bafilomycin A1, sensitize the tumor cells to therapeutic agents in colorectal, lung, esophageal cancer and leukemia and are currently under study in many clinical trials [72].
4) Metabolic disease
Autophagy is involved in metabolism of carbohydrate, protein, and lipid. Recent researches have suggested dysregulation of autophagy is implicated in several metabolic diseases such as diabetes, obesity, and atherosclerosis [73].
Dyregulation of autophagic activity in pancreatic β-cells was associated with insulin resistance and the pathogenesis of type 2 diabetes. Indeed, increased autophagosome formation was observed in mice β-cells in an insulin-resistant state caused by a high-fat diet and also ob/ob mice, a several diabetic rodents model [7475]. Since pancreatic β-cells are responsible for extensive proinsulin biosynthesis with higher burden of protein-folding, an increase of ER stress due to protein misfolding is also commonly occurred, inducing autophagy. It was suggested that activation of autophagy by increased ER stress in pancreatic β cells plays a protective role against cell dysfunction and death. This may be similarly applied to syncytiotrophoblast from placenta (will be discussed in Part II), which possess high burden of placental hormone biosynthesis, which can lead to excessive ER stress. It can be in this context that metformin, an antidiabetic drug, activates autophagy by inhibition of mTORC1 [76].
Intracellular lipids are stored in lipid droplets were known to be degraded by autophagy (lipophagy). Autophagy inhibition in cultured hepatocytes increased cellular triglycerides content and lipid droplet number and size in response to a lipid challenge [77]. It was demonstrated that excessive tissue lipid accumulation such as hepatic steatosis is caused by impaired lipophagy [78].
PART II
Autophagy in the placenta
1. Trophectoderm and early placenta development
Increased embryonic or fetal death caused by inactivation of autophagy-related genes in mouse models supports the essential roles of autophagy in embryo development. Tsukamoto et al. [13] created completely autophagy-deficient mice by using oocyte-specific Atg5-deficient mice. The oocytes lacking Atg5 were fertilized normally, but finally manifested embryonic lethality. Autophagy-defective oocytes derived from oocyte-specific Atg5 (autophagy-related 5) knockout mice failed to develop beyond the four- and eight-cell stages if they were fertilized by Atg5-null sperm, but they could develop beyond these stages if they were fertilized by wild-type sperm. Beclin 1-deficient mouse embryos have also been shown to die as early as embryonic day 7.5 [79]. Autophagy contributes to dead-cell clearance during programmed cell death by the generation of energy-dependent engulfment signals [80]. Impairment in the ability to clear apoptotic cells resulting in elevated inflammation may be responsible for embryonic lethality. Defective microautophagy due to the loss of either Rab7 or VPS41 function impaired gastrulation, the key developmental course by which animals establish the three germ layers [81].
Autophagy shows dynamic changes along the developmental stages of the embryo from activation right after fertilization [82]. Autophagy is activated in fertilized oocytes as early as 4 hours after sperm entry (one- to four-cell stage) through inhibition of the mTOR pathway and is believed to degrade superfluous maternal material and organelles from the fertilizing spermatozoon [1383]. For example, mammalian sperm mitochondria, located in the mid-position of the flagellum, enter the oocyte after gamete fusion and are degraded by autophagy [84]. The transition from oocyte to zygote involves many changes, including protein synthesis, protein and RNA degradation, and organelle remodeling [85]. The dynamic protein turnover by autophagy at this stage may be a critical determinant for normal embryonic development. In a study by Song et al. [86], treatment of bovine zygotes with rapamycin for transient elevation of autophagic activity during early preattachment development greatly increased the blastocyst development rate, trophectoderm cell numbers, and blastomere survival; these same parameters were reduced by both inhibition and prolonged induction of autophagy. After this initial induction during the transition from oocyte to zygote, there is a gradual decrease in autophagy-related gene transcription through the morula and blastocyst stages [79]. The reason for this downregulation of autophagy during this period remains unknown. It has been proposed that autophagy might be suppressed to prevent destruction of crucial factors for further embryonic development [87]. We speculate that suppression of autophagy during this period might be linked to preparation for implantation and invasion into the maternal uterus.
In mice, ovarian progesterone and estrogen are indispensible for the activation of blastocysts for implantation. In experimentally delayed implantation in mice, dormant blastocysts survive for more than 3 weeks in the absence of both estrogen and progesterone [88]. The precise mechanism that allows dormant blastocysts to prolong their viability appears to involve activation of autophagy, especially in trophectoderm, which may be programmed to conserve embryonic developmental function. Lee et al. [89] have shown that autophagy is induced when blastocysts are artificially maintained in a dormant state. It seems that blastocyst dormancy with autophagy activation in trophectoderm may result in impaired developmental fitness and may have long-lasting effects. The authors also have proposed that impaired ability to activate autophagy at later developmental stages may be a possible etiologic mechanism for development of fetal growth restriction.
The PI3K/Akt signaling pathway, which might be linked to the mTOR pathway, was reported to regulate the development of the differentiated trophoblast giant cell phenotype in a mouse model [90]. However, it has not yet been reported whether autophagy affects the differentiation of trophoblasts in human placenta. It can be speculated that, at different developmental stages, the cytotrophoblast and the syncytiotrophoblast may display distinctive patterns of autophagy. The role of autophagy in early placental development needs further exploration.
2. Autophagy and spontaneous abortion and the change according to gestation
Few studies investigated the change of autophagy in the placenta from spontaneous abortion, partially because retrieval of normal control samples cannot be obtained. Recently, a study by Avagliano et al. [91] demonstrated that enhanced expression of LC3-II in villous trophoblasts in spontaneous abortion with increased HIF-1α and cleaved caspase 3 in decidual tissue. Given the fact that the trophoblasts in human placenta have to adapt to the change in microenvironment at the maternal fetal interphase over nine months, gestational age-dependent change in placenta should not be underestimated. However, there is limited information on the change of autophagy over gestation. A study by Hung et al. [92] showed that advancing gestation was associated with decreased placental expression of LC3B-II and DRAM but not beclin 1. In our previous report, we could not find any significant difference in the expression of LC3-II or beclin 1 across the gestational period [93].
3. Autophagy in the placenta from women with preeclampsia and fetal growth restriction and its related pathway
We summarized the results from studies on autophagy in the placenta from women with preeclampsia and fetal growth restriction (FGR) (Table 1) [9394959697]. Several studies have demonstrated that placenta from women with preeclampsia or FGR showed increased autophagic activity in villous trophoblasts compared with placenta from a normal pregnancy. However, depending on the study population selected in each study, there are some conflicting points. Although some studies indicated that placentas from women with preeclampsia demonstrated increased autophagy regardless of the presence of FGR [94], other studies found that the increased autophagy in placenta was observed only in the group with preeclampsia complicated with FGR but not in the group with only preeclampsia [95]. Nonetheless, it is certain that the severity of preeclampsia or FGR is positively correlated with enhanced autophagosome formation. Therefore, it was suggested that increased autophagy in preeclampsia and FGR likely contributes to the development of FGR. However, it seems uncertain yet whether enhanced autophagy activity in trophoblasts from placenta is regulated at the transcriptional level because microarray data failed to show differential expression profiles of autophagy-associated genes in placentas from women with preeclampsia [98].
Recently, it was suggested that autophagy in trophoblasts is a protective mechanism against senescence, which is the irreversible arrest of cell growth and subsequent aging, and may participate in replacing damaged intracellular organelles such as ER and mitochondria during the recruitment of cytoplasm into syncytiotrophoblasts [99]. It was also demonstrated that unlike sera from normotensive women, autophagy induction in sera from women with preeclampsia was blunted, suggesting dysregulation of autophagy in the pathophysiology of preeclampsia [100].
Preeclampsia was suggested to be a disease of protein misfolding and aggregation [101]. Other diseases related to protein misfolding include neurodegenerative diseases such as Alzheimer disease and type II diabetes, which are well known to be associated with aberrant autophagic activity (accumulation of autophagosome in the target cells). Of note, neurons and pancreatic β cells, unlike most cells such as hepatocytes and muscle cells manifest less prominent starvation-induced autophagy [102]. We found that JEG-3 cells did not show a significant increase in LC3-II expression with rapamycin treatment (unpublished data).
What then mediates autophagy in villous trophoblasts in preeclampsia or FGR? As summarized in Table 2 [9293959697103104105106], in vitro hypoxia increased autophagy as assessed by the expression of LC3-II and p62 in primary human trophoblasts [95103]. Trophoblast cell lines including JEG-3, BEWO, and HTR8/SVneo cells also showed increased autophagosome formation in response to hypoxia [9396104]. As expected, an oxidative stress inducer (glucose oxidase) also enhanced autophagy in HTR8/SVneo cells [97]. Of note, it was also found that a p53 activator further increased autophagy in response to hypoxia, which was alleviated by a p53 inhibitor, indicating that p53 is involved in hypoxia-induced autophagy [95]. Collectively, it is evident that hypoxia enhances autophagic activity in primary trophoblasts or trophoblast cell lines.
So far, several mechanisms of how hypoxia induces autophagy have been investigated. As described earlier, different signaling pathways were activated depending on the degree of hypoxia or anoxia [29]. In brief, HIF-dependent signaling in moderate hypoxia (1% to 3% of oxygen) abates beclin 1/Bcl-2 interaction, resulting in autophagy, whereas an HIF-independent pathway in anoxia (<0.1% oxygen) induces autophagy via AMPK or the unfolded protein response pathway. Given the fact that placenta development at 8 to 10 weeks occurs under relatively hypoxic condition (<2% O2, 15 to 20 mmHg) [107], autophagy may play an important role in trophoblast invasion via an HIF-dependent signaling pathway. In fact, the exact molecular pathways of how hypoxia induces autophagy in trophoblasts are not yet clearly elucidated. Moreover, two studies regarding the effect of HIF1-α on autophagy in trophoblast cell lines (HTR8/SVneo cells) seemed to show conflicting results. One study showed that HIF1-α induced autophagy [105], whereas the other demonstrated that suppression of HIF1-α by siRNA increased expression of LC3 and autophagosome formation [108].
Nutritional deprivation, which is a potent autophagy inducer in other cells, was also assessed for autophagy in trophoblasts. Serum-depleted or glucose-free medium induced autophagic vacuolation or increased expression of LC3-II in BeWo cells and primary human trophoblasts, respectively. In our previous study, we found that TNF-α is a more potent autophagy inducer than hypoxic stimuli in JEG-3 cells and human term primary trophoblasts [93]. Interestingly, a recent study by Melland-Smith et al. [106] demonstrated that autophagy in JEG-3 cells is significantly triggered by ceramides, which are signaling molecules in the cellular response to stress and apoptosis. This group also showed that elevation of ceramides with decreased acid ceramidase was evident in placenta from preeclampsia compared with normal pregnancy and demonstrated that pharmacologic inhibition of acid ceramidase in pregnant mice induced abnormal placentation and reduced fetal growth as well as increased autophagy and placental ceramide content. Finally, they suggested that a shift of the Bcl-2-related ovarian killer-myeloid cell leukemia 1 rheostat toward pro-death Bcl-2-related ovarian killer may be implicated in preeclampsia.
Another possible candidate mechanism that mediates placental autophagy in a FGR pregnancy seems to be via the mTOR pathway, as mTOR is known to control the transfer of nutrients from mother to fetus [109]. Given the fact that FGR is associated with reduced activity of placental mTOR [110], a potent inhibitor of autophagy, enhanced autophagy in FGR pregnancies makes sense in this context. In addition, it was also demonstrated that hypoxia (<1% oxygen) inhibited mTOR1 activity in primary trophoblasts as assessed by decreased expression of phosphorylated ribosomal protein S6 [103].
4. Autophagy and trophoblast invasion
Trophoblast invasion is regulated by various cellular processes, including apoptosis, proliferation, and differentiation [111]. As there is a close relationship between trophoblast apoptosis and invasion, regulation of trophoblast invasion by autophagy is an important subject to be studied.
Conflicting findings regarding the role of autophagy in cellular invasion have been reported depending on cell types and experimental conditions such as autophagy inhibition or induction methods [112]. Inhibition of autophagy limited cellular invasion of glioblastoma and hepatocellular cancer cell lines [113]. In contrast, increased autophagic activity was linked to less tumor invasion in breast cancer cells and in tongue squamous cell carcinoma cells [114115].
Despite the critical importance of trophoblast invasion in pathologic pregnancy including preeclampsia and FGR as well as normal pregnancy, few studies have assessed trophoblast invasion in relation to autophagic activity, and they showed somewhat conflicting results. A study by Yamanaka-Tatematsu et al. [105] demonstrated that CoCl2 with increased HIF1-α suppressed trophoblast invasion in autophagy-deficient HTR8/SV cells in which suppression of matrix metalloproteinase-9 activity and decreased cellular ATP levels were involved. This research group also showed that the invasion and vascular remodeling assessed by tube formation was significantly diminished in autophagy-deficient HTR8/SV cells. Notably, soluble endoglin, which is abundant in sera of women with preeclampsia, was found to suppress the invasion of extravillous trophoblasts by inhibiting autophagy [104]. Recently, it was demonstrated that expression of p62, a marker of impaired autophagy, in extravillous trophoblasts in the decidua basalis was significantly increased in preeclamptic compared with normotensive women [116]. In contrast, Choi et al. [108] revealed that HIF1-α inhibition by siRNA decreased trophoblast invasion along with increased autophagosome formation in HTR8/SV cells. Notably, in this study, the association between increased autophagy and reduced invasion was observed in both normoxic and hypoxic conditions. Another study by Hung et al. [92] showed that inhibition of autophagy by siRNAs against beclin 1, DRAM, and LC3B did not affect trophoblast invasion or cell viability in JEG-3 cells.
In our experiments, we inhibited autophagy in both JEG-3 and HTR8/SV cells by specific suppression of autophagy with beclin siRNA and short hairpin RNA and found that the invasiveness of these trophoblast cell lines was significantly increased when autophagy was inhibited (manuscript in preparation). Interestingly, we also observed that inhibition of autophagy in these trophoblast cell lines was associated with enhanced nuclear factor-κB activity.
5. Autophagy and apoptosis in trophoblasts
It is evident that increased trophoblast apoptosis was observed in placenta from preeclampsia and/or FGR pregnancies [117118119]. Considering the complex cross-talk between autophagy and apoptosis, there should be inter-regulation of autophagy and apoptosis in trophoblasts with respect to the formation of placenta and maturation throughout human gestation. Unfortunately, there are scant data for addressing this topic. A study by Chen et al. [103] demonstrated that silencing Atg7, an important enzyme in elongation of preautophagosomal structures, decreased both apoptosis and LC3-II in primary cultured trophoblasts. In line with this study, we observed that inhibition of autophagy by knockdown of LC3 or beclin 1 attenuated TNF-α-induced apoptosis in JEG3 cells and primary cultured trophoblasts [120]. We also observed that inhibition of autophagy repressed TNF-α-induced expression of tAtg5, which was reproduced by transfection with calpain siRNA, suggesting that cross-talk between autophagy and apoptosis might be regulated by calpain-mediated production of tAtg5.
However, considering multiple crossroads, our understanding of the interconnection between autophagy and apoptosis seems to be at an early stage, and therefore further extensive investigations are urgently required in human trophoblasts.
6. Autophagy and parturition: term and preterm
The possible association between autophagy and term parturition has been investigated in only a few studies. However, the limited evidence leans toward the finding of possible suppression of autophagy in association with spontaneous labor. A study by Signorelli et al. [121] indicated that autophagy in placenta was significantly higher in the no-labor group (i.e., they underwent elective cesarean section owing to breech presentation) compared with the spontaneous labor group. Reduced capacity of autophagy along with a polymorphism in the gene coding Atg16L1 was also reported to be associated with spontaneous labor [122]. No difference in LC3-II expression in placentas from spontaneous versus medically induced labor has been reported [123]. Interestingly, this study also reported a significant association between placental LC3-II expression and maternal prepregnancy body mass index. A recent study by Brickle et al. [124] showed decreased expression of autophagy proteins (beclin 1, Atg3, Atg5, Atg7, Atg12, and Atg16L1) in fetal membranes after spontaneous onset of labor in term and preterm birth and preterm premature rupture of membranes. They also showed that an autophagy inhibitor (LY290042) enhanced lipopolysaccharide-induced IL-1β secretion from fetal membranes, suggesting the activation of autophagy as a therapeutic mechanism to delay infection-induced preterm birth.
There are limited data on autophagy in gestational tissue in relation to preterm parturition. Strong LC3 expression in inflammatory cell infiltrates in the fetal membrane has been demonstrated, but LC3 expression in trophoblasts from chorionic laevae was decreased in preterm delivery with histologic chorioamnionitis compared with delivery without inflammation [125]. Interestingly, an animal study using pregnant mice demonstrated that inhibition of autophagy by heightened mTORC1 signaling resulted in preterm birth caused by premature senescence in decidual cells [126]. A recent study by Agrawal et al. [127] showed that the autophagic flux was altered in a mouse model of inflammation-induced preterm birth, as reflected by increased LC3B expression and decreased Atg4, Atg7, lysosomal-associated membrane protein (LAMP)-1, and LAMP-2 expression in uterus and placenta.
Taken together, considering that parturition, whether preterm or term, is itself an inflammatory process, reduced autophagy activity contributes to inadequate clearance of inflammasomes, resulting in excessive proinflammatory cytokine release and triggering parturition [128]. Of note, the above studies regarding autophagy and parturition concomitantly suggest that at least the presence of labor or the mode of delivery (vaginal or cesarean delivery) should be controlled when comparing autophagy in placentas from different groups.
7. Autophagy and antiviral role of placenta
In placenta, trophoblasts perform the role of a guard at the maternal-fetal interface throughout pregnancy. One of the critical roles of trophoblasts is the immunologic barrier to protect the fetus from the invasion of microorganisms threatening fetal life. Recently, a study by Delorme-Axford et al. [129] presented that exosomes derived from primary trophoblasts contained placenta-specific microRNAs (miRNAs) (C19MC), which induced autophagy and exerted antiviral immunity at the maternal-fetal interface. Accordingly, it was implicated that exosomes secreted from trophoblasts confer antiviral resistance to nearby cells by autophagy-inducing miRNAs. In fact, after the first report by Zhu et al. [130] stating that miR-30a could negatively regulate autophagic activity in cancer cells, accumulating evidence has indicated that many miRNAs are involved in the control of autophagic activity [131].
Conclusion
In the past few decades, there have been substantial efforts to search for new knowledge about autophagy, which was observed and termed approximately a half century ago. Au-tophagy is primarily considered an adaptive response for cell survival to defend against intracellular or extracellular stress such as nutritional deprivation, hypoxia, ER stress, mitochondrial damage, infection, and inflammatory signals. Multiple upstream signaling pathways and cross-talk with apoptosis have been actively explored.
In relation to human pregnancy, several important studies on autophagy in the placenta are now being reported. Autophagy has an indispensable role in early embryo development. Placentas from preeclampsia and FGR are associated with increased placental autophagosome formation. As for the underlying mechanisms of increased autophagy in these pregnancy complications, several stimuli including hypoxia, increased ER stress, reduced mTOR activity, inflammatory stress, and elevation of ceramides in trophoblasts have been suggested so far. Several findings have also indicated that autophagy affects trophoblast invasion, but there are conflicting results depending on cell types and experimental conditions. Interestingly, recent studies suggested that human parturition, whether preterm or term, was associated with reduced autophagic activity. Autophagy was also identified to have an antiviral role in placenta by forming miRNAs in exosomes. Considering the essential role of the placenta in maintaining a healthy pregnancy and the basic concept of autophagy as an adaptive response to various stressed conditions, more extensive research on autophagy in the placenta are challenging and expected.


 XML Download
XML Download