

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Polycystic ovary syndrome (PCOS) is one of the most common endocrine disorder affecting 5% to 10% of reproductive aged women [1]. PCOS is a heterogeneous disorder with a broad spectrum of clinical manifestations and now well recognized as having a major effect on the reproductive, metabolic and cardiovascular function of affected women throughout life [2]. Although the pathophysiology of PCOS remains incompletely understood, this condition is known to be a multifactorial complex condition with a strong genetic component that is associated with ovulatory dysfunction, insulin resistance and hyperandrogenism [2].
Hyperandrogenism is one of the characteristic features of PCOS. Therefore, polymorphism of genes involved in androgen signaling are considered to be related to the development of PCOS. Androgen action is mediated by the androgen receptor (AR), and the gene encoding AR that is located in the Xq11-q12 region contains highly polymorphic CAG trinucleotide repeat region in exon 1 [3]. The expansion of CAG repeat is known to affect the AR activities, and CAG repeat number of AR gene is inversely correlated with the expression and transactivation potential of AR [3]. AR polymorphism has been reported to be related to the development of PCOS or hyperandrogenism [4,5,6,7]. AR gene polymorphism has been also associated with other forms of androgen pattern diseases including androgen insensitivity syndrome (AIS) [8]. The AIS is a X-linked recessive disorder which occurs in one out of 20,000 births. Patient with AIS presents female phenotype with normal male karyotype because of the partial or complete unresponsiveness of the cells to androgen. Brown et al. [8] first reported that mutations in the AR gene located in the X chromosome are responsible for the AIS. Almost 70% of affected individuals inherit the mutation from their carrier mother [9]. Both PCOS and AIS seems to have a similarity in abnormal androgen activity due to possible AR gene polymorphism. In this case report, we present a PCOS woman with heterozygous AR gene mutation who gave birth to a baby with complete AIS.
Case report
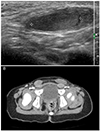
A 37-year-old Korean woman (gravida 3 and para 1) with birth history of the child with AIS was referred to our Reproductive Medicine and Infertility Clinic for infertility treatment. Her body mass index was 22.1 and Ferriman-Gallway index score for diagnosis of hirsutism was 5. She had no growth or sexual developmental abnormalities, or abnormal gynecological symptoms except oligomenorrhea with an interval of 35 to 50 days. She did not have any particular family history including diabetes and cardiovascular disease. In 2008, she was naturally pregnant and delivered a baby girl at term by cesarean section. Eight months after birth, walnut sized soft masses were palpated at both inguinal area of her baby, and the baby had undergone a herniorrhaphy under the diagnosis of bilateral inguinal hernia. In an operating room, testis- and spermatic cord-like masses were accidentally found in both inguinal canals. Therefore, this baby was referred to our Medical Genetic Center for diagnostic workup. The external genitalia showed grossly normal appearance of a baby girl. A scrotal ultrasonography (USG) and abdomino-pelvic computed tomography (CT) scan were performed, and both testes were found in both inguinal canals (Fig. 1). The uterus, vagina and ovaries were not identified in the abdomino-pelvic CT scan (Fig. 1). Under the impression of complete AIS, cytogenetic analysis for her baby was performed, and the karyotyping showed 46,XY, inv(9) (p12q13). Therefore, this baby was diagnosed as complete AIS. Her testes are scheduled to be checked regularly and removed immediately after puberty.
Genotyping for the AR gene polymorphism of the patient (mother of AIS baby) and her AIS baby was performed. DNA was extracted from peripheral leukocyte using a standard phenol-chloroform extraction method. Partial sequence of the proband eight exons and their exon-intron boundaries of AR gene was sequenced using BigDye terminatore v.3.0 and ABI3130xl Genetic Analyzer (Appliedbiosystems, Forster City, CA, USA) with genomic DNA isolated from peripheral leukocyte. In AIS baby, homozygous c.2482T>C (p.Phe828Leu) mutation on exon7 was detected (Fig. 2). This proband was strongly suggestive of AIS patient from the variation of this mutation. In the µdetected and it was consistent with carrier of AIS (Fig. 2).
Routine workup for infertility and oligomenorrhea was performed in our Reproductive Medicine and Infertility Clinic. On the 3rd day of menstruation, her blood was drawn for hormonal assay. Serum levels of follicle stimulating hormone, luteinizing hormone, prolactin, thyroid stimulating hormone, thyroxine, 17α-hydroxyprogesterone, cortisol, insulin-like growth factor-1, dehydroepiandrosterone, glycosylated hemoglobin, and insulin were 2.9 mIU/mL, 2.1 mIU/mL, 4.6 ng/mL, 0.63 µU/mL, 1.2 ng/dL, 0.96 ng/mL, 13.9 µg/dL, 104 ng/mL, 924 ng/mL, 5.2% and 2.3 µIU/mL respectively, all of which were within normal ranges. However, serum antimüllerian hormone level was 9.42 ng/mL, which was in 90 to 100 percentile of reproductive aged women. Also, serum testosterone and free testosterone concentrations were 0.58 ng/mL and 0.85 pg/mL respectively, which represented hyperandrogenemia in female based on the reference values of our institute. Transvaginal USG performed on the 7th and 14th day of menstruation showed a normal retroverted uterus and both ovaries with polycystic ovary morphology. Both right and left ovaries contained 15 follicles measuring 2-9 mm in diameter per each ovary and increased ovarian volume of 12.5 and 10.8 mL, respectively. Dominant follicle more than 10 mm in a mean diameter, corpus luteum or abnormal cyst was not identified. Doppler USG on the intraovarian arteries revealed decreased resistance index of 0.48 in each ovary. Therefore, she was diagnosed as a PCOS based on the revised PCOS diagnostic criteria of the 2003 Rotterdam consensus [10]. She is currently getting in vitro fertilization treatment for preimplantation genetic diagnosis.
Discussion
PCOS is a multifactorial heterogenous disorder with a strong genetic component that is characterized by hyperandrogenemia and/or insulin resistance, ultimately leading to ovulatory dysfunction including amenorrhea and oligomenorrhea, polycystic ovarian morphology and infertility [1,2]. Although its pathophysiology is not fully understood, androgen excess is considered to be an important feature of PCOS and arise primarily from dysregulated ovarian androgen secretion. We still do not know clearly what brings hyperandrogenic status in this disorder, but AR gene polymorphism has been reported to be a promising association factor and biomarker for PCOS or ovarian androgen excess [4,5,6,7].
In AR gene located in the Xq11-12, protein coding region (about 2,757 Kp open reading frame) comprises 8 exons, designated A-H or 1-8, separated by introns up to 26 kb in size. Exon 1 encodes the amino-terminal domain which comprises more than half of the AR protein. Toward the 5'end of exon 1 is a CAG repeat region (polyglutamine, PolyQ) that contains a mean repeat number of 21±2, and this repeat region is highly polymorphic [3]. Especially AR CAG repeat length may be important in the etiology of PCOS, and it seems to contribute to individual differences in the susceptibility to the development of PCOS. In general, AR polymorphism might not be a major risk factor for PCOS. However, in the present case, heterozygous c.2482T>C (p.Phe828Leu) mutation of AR gene is thought to be a possible etiologic factor or one of etiologic factors of PCOS, because this AR gene mutation transmitted from PCOS mother resulted in an AIS by androgen resistance in her baby and any other risk factors for PCOS were not found in this patient. That is to say that c.2482T>C (p.Phe828Leu) mutation of AR gene can be a cause of androgen resistance in AR and PCOS and it is proven by an AIS baby with AR gene mutation inherited from this patient. Therefore, not only CAG repeat length variation but also point mutation in AR gene can may be a contributing genetic factor for PCOS.
AR gene polymorphism is also associated with the development of different androgen pattern disease including Kennedy's spinal and bulbar muscular atrophy [11]. AIS characterized by loss of AR function and variable defects in virilization of 46, XY individuals can be also caused by AR gene mutations [8]. Since AIS, so called testicular feminization was first described by Morris in 1953 [12], underlying causes have been explored. AIS is associated with various molecular defects including point mutations resulting in amino acid substitution or premature stop codon, alteration of CGA repeat number, complete or partial gene deletions and intronic mutations. Recently, Werner et al. [13] documented experimentally the contradictory effect of the combination of a short polyglycine repeat with the rare mutation of the hinge region A645D, resulting in seriously reduced AR activity when paired with a long PolyQ (CAG repeat) repeat and almost wild-type AR activity when paired with a short PolyQ repeat [13]. In this case, homozygous c.2482T>C (p.Phe828Leu) mutation on exon7 was identified from the AIS baby (Fig. 2). This mutation has not been reported for AIS, but this mutation is most likely to be a cause of AIS. This mutation of an affected baby was thought to be inherited from her carrier mother with PCOS. Actually, identified mutations of the AR gene may be recurrent, and approximately 70% of AIS patients inherit the mutation [9]. Whereas about one third are developed by de novo germline or somatic mutations.
In gynecological endocrinologic field, we can encounter a variety of endocrine disorders representing hyperandrogenism. Gynecological endocrinopathies characterized by hyperandrogenism such as PCOS can share the specific molecular defects like AR mutations with other forms of androgen pattern disease such as AIS. This case demonstrated that in PCOS patients with hyperandrogenism caused by AR gene mutations, their genetic defects can be inherited to their offspring and lead to the development of AIS. The present case suggests that the heritability of AR gene mutations in PCOS patients should be considered despite a rare possibility of comorbidity of AIS carrier and PCOS.

 XML Download
XML Download