

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Congenital leukemia (CL) is a very rare disease that is seen from 1 to 5 per million live births, which develops in utero and usually is diagnosed at birth or within one month of life because of the rapid doubling time of leukemic cells [1,2]. It is a unique disease that most of them are of myeloid origin in contrast to general pediatric leukemia, which are usually lymphoid in origin [3]. Clinical manifestations of CL are similar to infectious diseases, therefore it is obscurant to make a precise diagnosis, for which further evaluation and diagnostic tools are necessary [4]. Antenatally, diagnosis of CL cannot be made easily due to high risk of fetoscopy or cordocentesis, but fetal hepatosplenomegaly observed by ultrasonography can provide a clue for diagnosis of CL. Fetal hepatosplenomegaly may be presented in various conditions, most commonly in Rh isoimmunization and other forms of fetal anemia, congestive heart disease, infectious disorders such as cytomegalovirus, metabolic disorders such as Gaucher disease or pyruvate kinase deficiency, and transient myeloproliferative disease (TMD) which sometimes appears in fetuses with Down syndrome and overgrowth disorder [5]. In this report, we are to present a case of an incidental hepatosplenomegaly of fetus complicated with acute myeloid leukemia associated with AML1 gene duplication, which was diagnosed by bone marrow aspiration for identification of the cause of a newborn's leukemoid reaction.
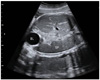
Case report
A 36-year-old woman, had undergone in vitro fertilization and embryo transfer, was referred to our institution for evaluation of fetal hepatosplenomegaly at 37 weeks' gestation. The index pregnancy was otherwise uncomplicated. Her past history was uneventful, and the results of antenatal laboratory tests were normal. A fetal heart rate was about 150 beats per minute. Fluctuations in the fetal heart rate are called variability were normal without distress sign. A transabdominal ultrasonography with 3.5-5 MHz transducer (Voluson 730 pro, GE, Seoul, Korea) revealed a single, live fetus with measurements corresponding to the gestational age except abdominal circumference, due to hepatosplenomegaly. Findings of fetal hydrops also were not observed. A total amniotic fluid index measured 17.5 cm which is normal range. Hepatomegaly means that the liver reaches into the fetal pelvis adjacent to the fetal bladder. Length of liver in this period is normally varies from 40 to 60 mm. It can be measured from the dome of the diaphragm to the tip of the right lobe parallel to the long axis of the fetal trunk [6]. Splenomegaly is diagnosed by three findings. First, the stomach is displaced anteriorly and to the midline by the enlarged spleen. Second, the spleen is very close to the anterior abdominal wall. Third, color Doppler imaging depicts multiple ramifications of the splenic artery and vein. Length of spleen in this period is normally measured from 45 to 60 mm [7]. Echogenecity of both organs is usually isoechogenic. However, in this case, size of liver and spleen were above 95th percentile of the upper limit and also the Fig. 1 shows both organs adjacent to bladder, which indicates fetal hepatosplenomegaly. Although limited clinical information was available to discuss the ideal mode of delivery with the parents, we recommended cesarean delivery next day to prompt evaluation of rapid progression of fetal hepatosplenomegaly. At delivery, the newborn, a male, weighted 3,070 g was born and Apgar scores were 5 and 7 at 1 and 5 minutes, respectively. When the newborn was physically examined, it revealed unstable respiration with tachypnea and desaturation due to abdominal distension. Intubation was immediately performed and the baby was mechanically ventilated in the neonatal intensive care unit. The abdomen was moderately distended and liver and spleen were palpable 4 and 2 cm below the costal margins. No skin lesions were present (e.g., twenty five to thirty percent of infants with CL have specific cutaneous infiltrates; leukemia cutis which usually appear as firm blue or red nodules called Blueberry Muffin) [8].
On laboratory tests performed at birth, the hemoglobin concentration was 13.3 g/dL, hematocrit was 42.9%, platelet cells count was 228×103/UL, and white blood cells count was 28.18×103/UL. A differential cell count revealed 25% neutrophils, 44% lymphocytes, 4% monocytes, 3% eosinophils, 0% basophils, 1% myelocytes, and 23% atypical lymphocyte. Few atypical cells were observed on peripheral blood smear and initial peripheral blood chromosome study reveals normal karyotype. Also, serology studies for TORCH syndrome, including serum antibody titers against toxoplasmosis, cytomegalovirus, rubella, and herpes simplex virus and venereal disease research laboratory tests were all negative ruling out infectious condition. Another work-up for septic condition and tandem mass for metabolic disorder were all negative. While further evaluation and monitoring were performed, aspects of hepatic failure were consistently shown with elevated total bilirubin (7.38 mg/dL) in spite of exchanged transfusion for prevention of kernicterus. On peripheral blood test performed on 28 days after birth, the hemoglobin concentration was 10.7 g/dL, hematocrit was 33.4%, platelet cells count was 123×103/UL, and white blood cells count was 67.94×103/UL. A differential cell count revealed 20% neutrophils, 6% lymphocytes, 22% monocytes, 19% eosinophils, 3% basophils, 0% myelocytes, and 30% atypical lymphocyte. Increased numbers of atypical cells which are large-sized immature blasts were seen in a peripheral blood smear (Fig. 2A), therefore bone marrow aspiration was performed. A bone marrow aspirate smear showed a moderately cellular marrow with abnormal proliferation of immature blasts (≥35%) and granulocytic precursors and negative for myeloperoxidase, Sudan black B and periodic acid Schiff stain. The erythroid precursors and megakaryocytes were decreased in number (Fig. 2B). Immunophenotyping revealed that the 75% of blasts were positive for myeloid leukemic marker of CD33+. The final diagnosis was acute myeloid leukemia. Work-up for detecting metastasis of leukemic cell was done, and there were no specific finding except small ventricular septal defect in computed tomography angiography.
To figure out the cause of CL, especially for recurrent genetic abnormalities in de novo AML, fluorescence in situ hybridization (FISH) for core-binding factor subunit beta, myeloid lymphoid lineage, AML1/ETO gene rearrangement were done. Although recurrent genetic abnormalities were not detected, 94% of three AML1 gene signals which could be interpreted as trisomy 21 were detected in this bone marrow specimen. The AML1 (CBFA2) gene, located in the chromosomal band 21q22, has recently attracted a lot of interest in terms of its role in leukemogenesis [9]. Retrospectively, to investigate the three AML1 signals on initial peripheral blood specimen shown normal karyotype, we performed the FISH, AML1/ETO gene rearrangement. FISH results showed 50% three AML1 signals on initial peripheral blood specimen. Based on the presence of three AML1 signals with normal karyotype in initial study, this patient was thought to have a clonal change of cryptic three AML1 gene signals carrying normal karyotype, and in other words, this result can confirm the presence of CL with AML1 gene duplication. Three AML1 clonal progressions were noted in bone marrow specimen (Fig. 2C). Therefore, this patient had very low possibility of Down syndrome except the case of very low level of mosaic Down syndrome.
Even though the infant was treated with chemotherapy (idarubicin, cytarabine, etoposide, and daunorubicin), ventilator care and inotropic agents, the baby developed increasing respiratory distress and hypotension. He died of respiratory distress with pneumonia at 6 weeks after birth.
Discussion
CL becomes clinically evident at birth or within the first month of life because of the rapid doubling time of leukemic cells. It is a rare entity, with reported incidence that ranges from 1 to 5 per million live births [1,2].
To fulfill the criteria of CL, first, there must be manifest at or shortly after birth(<30 days) and second, the blood should reflect an alteration of the bone marrow by showing the presence of immature cell proliferation of myelomonocytic, lymphoid, or erythroid series or in extrahematopoietic tissue. Third, there should be absence of any other condition that might cause leukemoid reactions mimicking CL, such as, congenital infections (TORCH), hemolytic disease of the newborn (HDN), hereditary spherocytosis, twin to twin transfusion, other neoplastic infiltrates and TMD, a nonmalignant leukocytosis seen usually in neonates with Down syndrome [8,10]. Therefore, to perform accurate diagnosis, differential diagnosis is very important. In this case, the infant could be diagnosed as congenital leukemia satisfying all three conditions mentioned above.
The causes of CL are still unknown, but it has been associated with exposure to radiation, dietary compounds called bioflavonoid, use of tobacco and illicit drugs, and other substances, as well as coexisting congenital anomalies and the possibility of an underlying genetic predisposition in infants born with leukemia [10]. In this case, this patient was thought to have a clonal change of cryptic three AML1 signals carrying normal karyotype. Therefore, this result can confirm the presence of CL with AML1 gene duplication.
CL is often associated with hepatosplenomegaly, petechia, ecchymosis, lethargy, pallor and poor feeding at or shortly after birth and most infants die of respiratory distress which is a result of pulmonary leukostasis and bronchopneumonia [11]. In this case, there was no specific skin lesion found except hepatosplenomegaly at birth and the baby died of respiratory distress cause by pneumonia. Furthermore, autopsy was needed for diagnosis of pulmonary leukostasis and bronchopneumonia. However, it could not be done due to parents' rejection.
The differential diagnosis should include sepsis, intra-uterine infections (TORCH), HDN and TMD which is seen usually in association with Down syndrome. Historically, TMD has been described as a benign condition association with transient polycythemia and thrombocytosis, because the leukocytosis and its secondary clinical complications typically disappear without treatment over the first 2 months of life [4,10]. In this case, infectious diseases were ruled out through serologic studies and negative bacterial cultures, and HDN was also excluded through decreased numbers of erythroid precursors in peripheral blood. In addition, TMD which sometimes appears in fetuses with Down syndrome was excluded through identifying the presence of three AML1 signals carrying normal karyotype.
Once the diagnosis of leukemia is established, an intensive multi-agent chemotherapeutic regimen should be instituted. There is no specific therapeutic protocol for the treatment of either neonatal acute lymphoid leukemia or AML. Whether they undergo chemotherapy or not, neonates with leukemia usually require treatment in an intensive care unit [12].
CL has a poor prognosis, with only 23% surviving at 24 months [13,14]. However, a few patients were described that they have undergone spontaneous remission of CL without chemotherapy [15].
In conclusion, if during perinatal period, hepatosplenomegaly, hydrops and polyhydramnios by ultrasonography can be detected, perinatologist should consider various possibilities such as infectious disease, hemolytic disorder, metabolic disorder and TMD associated with Down syndrome. If it is possible, fetoscopy and umbilical blood sampling, which are not preferred to be performed, is needed. Finally perinatologist can then establish the diagnosis of leukemia beyond performing differential diagnosis.
We would like to report this case, which shows the fetus with the features of hepatosplenomegaly during perinatal period, on 37 weeks' of gestational age, and being diagnosed as CL associated with acquired AML1 gene duplication after birth through bone marrow aspiration.

 XML Download
XML Download