

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Preeclampsia is one of the leading causes of maternal mortality/morbidity and preterm delivery in the world, affecting 3% to 5% of pregnant women [1,2]. It is a syndrome defined by the onset of hypertension (≥40/≥90 mm Hg) and proteinuria (≥0.3 g/24 hr) after 20 weeks of gestation. Preeclampsia is the leading cause of maternal mortality in developing and developed countries. Delivery of the placenta remains the only known treatment for this disease, suggesting that the placenta is the principal contributor to the pathogenesis of preeclampsia [3].
The pathophysiology of preeclampsia likely involves both maternal and fetal/placental factors. Abnormalities in the development of placental vessels early in pregnancy may result in placental hypoperfusion, hypoxia, or ischemia. However, the molecular mechanisms behind preeclampsia are not clear and the role of angiogenic proteins in early placental vascular development is under investigation.
Abnormal Development of the Placenta
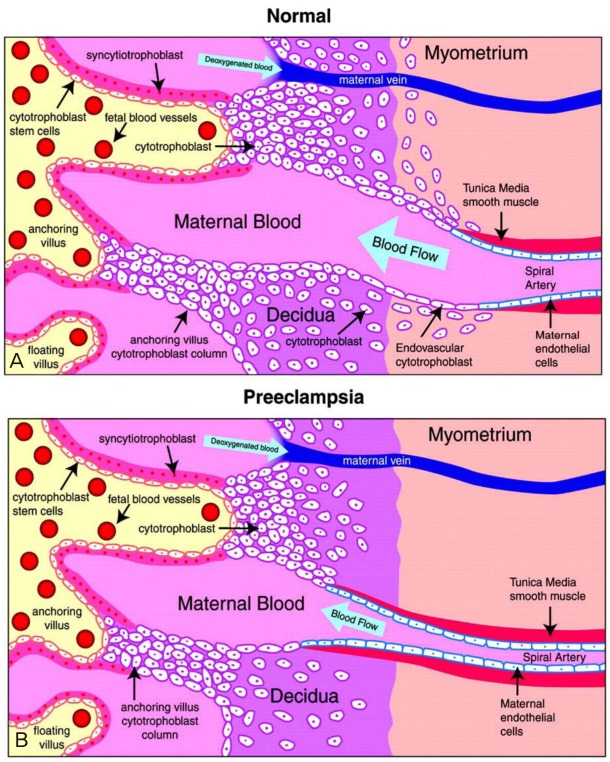
Because the placenta is central to preeclampsia pathogenesis, this research has focused on the association between abnormal placental vascular development and the development of preeclampsia. In normal pregnancies, extravillous cytotrophoblasts of fetal origin invade the uterine spiral arteries of the decidua and myometrium [3]. These invasive cytotrophoblasts replace the endothelial layer of the maternal spiral arteries, transforming them from small, high-resistance vessels into large-caliber vessels. However, in preeclampsia, this transformation is incomplete [4]. Cytotrophoblast invasion of the spiral arteries is limited to the superficial decidua and does not reach the myometrium (Fig. 1) [4,5].
Defective differentiation of trophoblasts might be an explanatory mechanism for defective trophoblast invasion of the spiral arteries in preeclampsia. For trophoblast differentiation to occur there must be alterations in the expression of a number of different classes of molecules, including cytokines, adhesion molecules, extracellular matrix molecules, metalloproteinases, and the class IB major histocompatibility complex molecule, histocompatibilty leukocyte antigen-G [6]. During normal pregnancies, invading trophoblasts alter their adhesion molecule expression from one set of endothelial transmembrane receptors (integrin alpha6beta1, integrin alphavbeta5, and E-cadherin) to another (integrin alpha1beta1, integrin alphavbeta3, and VE-cadherin) [7]. Hypoperfusion, hypoxia, and ischemia are critical components in the pathogenesis of preeclampsia because the hypoperfused placenta elaborates many factors into maternal vessels that alter maternal endothelial cell function and lead to the systemic symptoms of preeclampsia [8-10].
Altered Angiogenic Balance and Circulating Angiogenic Factors
Many angiogenic factors are produced by the human placenta. The most important factors are vascular endothelial growth factor (VEGF) and placental growth factor (PlGF). VEGF is an endothelial-specific mitogen that plays a key role in promoting angiogenesis [11]. VEGF's activities are mediated by its interaction with two high-affinity receptors-tyrosine kinase-kinase-insert domain region (the kinase domain region or vascular endothelial growth factor receptor-2) and fms-like tyrosine kinase 1 (flt-1). PlGF is an angiogenic growth factor that is thought to amplify VEGF signaling by displacing VEGF from the flt-1 receptor and allowing it to bind to the more active kinase-insert domain (KDT) receptor [11]. Increased sFlt-1 during preeclampsia is associated with decreased free VEGF and free PlGF in the blood [5]. Also, administration of sflt-1 to rats resulted in elevated blood pressure and proteinuria, indicating that excessive placenta-derived sflt-1 contributes to preeclampsia [12]. Soluble endoglin is an antiangiogenic protein, which acts together with sflt-1 to induce a severe preeclamptic-like syndrome in pregnant rats. Circulating soluble endoglin levels increased markedly beginning from two to three months before the onset of preeclampsia [11]. An increased level of soluble endoglin was associated with an increased endoglin/sflt-1 ratio [13].
There is substantial evidence that effective angiogenesis requires the synthesis of bioactive endothelium-derived nitric oxide (NO) [14]. A number of angiogenic factors up-regulate the endothelial expression of NO synthase (NOS) stimulating the release of endothelium-derived NO. NOS is partially regulated by negative feedback from NO, but there are other important inhibitors that are endogenously present in humans, such, the competitive inhibitor asymmetric dimethylarginine (ADMA) [15]. There are some conflicting findings concerning ADMA concentrations in pregnancies complicated by preeclampsia. However, Kim et al. [16] suggested that increased maternal circulating ADMA levels, a higher expression of placental eNOS protein, and a lower expression of placental phospho-eNOS protein contribute to the development of preeclampsia.
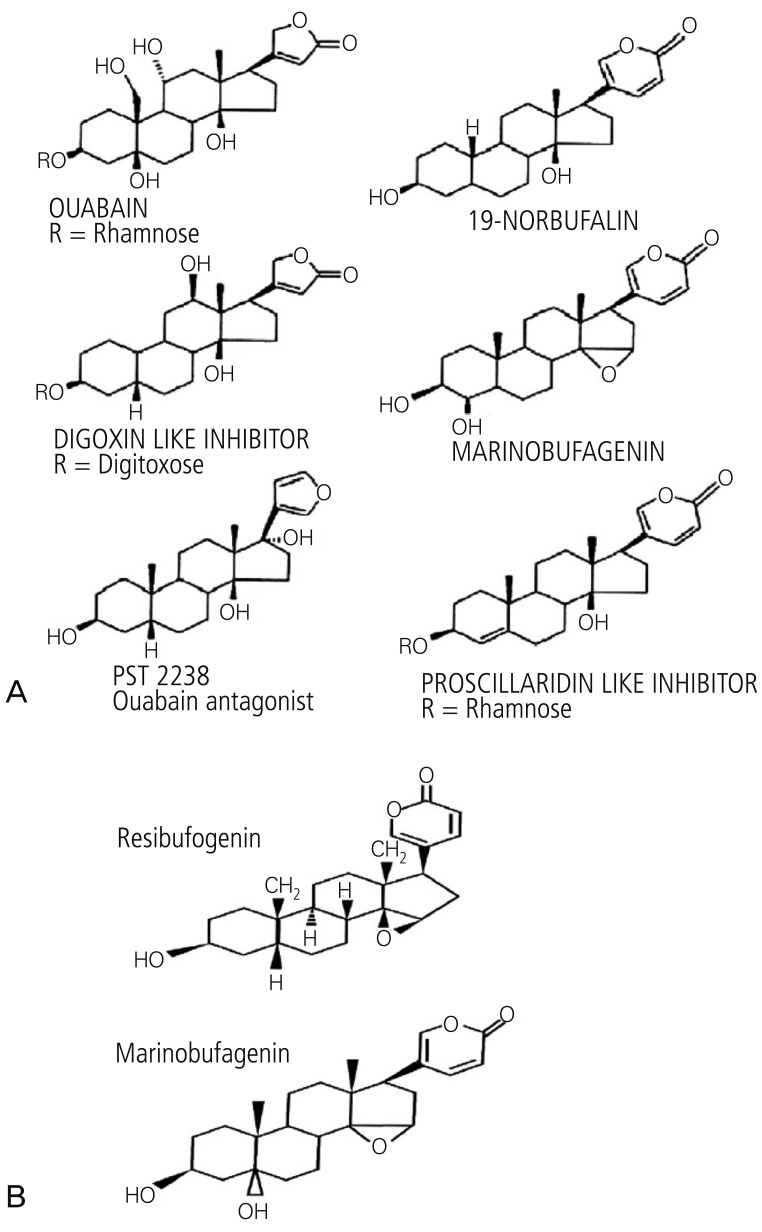
Marinobufagenin, a Bufadienolide Trigger of Preeclampsia Pathogenesis
Presently, the key biomarkers of the syndrome associated with preeclampsia pathogenesis are thought to be marinobufagenin (MBG) and angiogenic imbalance [17]. Data from a rat model that mimics human preeclampsia showed that urinary excretion of MBG increased before the onset of hypertension and proteinuria, and that affected animals have increased vascular leakage and blood-brain barrier permeability [17]. The cardenolides and bufadienolides are group of steroid compounds, which belong to a class of circulating substances called "cardiac glycosides" (Fig. 2) [18,19]. Cardiac glycosides have the ability to inhibit the sodium transport enzyme, sodium/potassium adenine triphosphatase [17]. They are also natriuretic and cause vasoconstriction, which can lead to hypertension as an agent of cardiac inotrope [20]. The bufadienolides are more important in diseases such as preeclampsia than the cardenolides [21,22]. The best studied of the bufadienolide compounds is MBG (Fig. 2A), an endogenous vasoconstrictor mammalian cardiotonic bufadienolide [18]. Preeclampsia is thought to result from inadequate placentation, related to a failure of the trophoblasts to adequately remodel the vasculature of the uterus [23]. This leads to hypoperfusion of the placenta, often causing oxidative stress, and endothelial dysfunction, which are related to the development of the preeclampsia syndrome [17]. MBG interferes with proliferation, migration, and the invasive capacity of the cytotrophoblasts [23], and has deleterious effects on human cytotrophoblast cell function, which suggests a role for MBG in the abnormal placentation and altered vascular function of preeclampsia.
Activation of the Renin-Angiotensin System
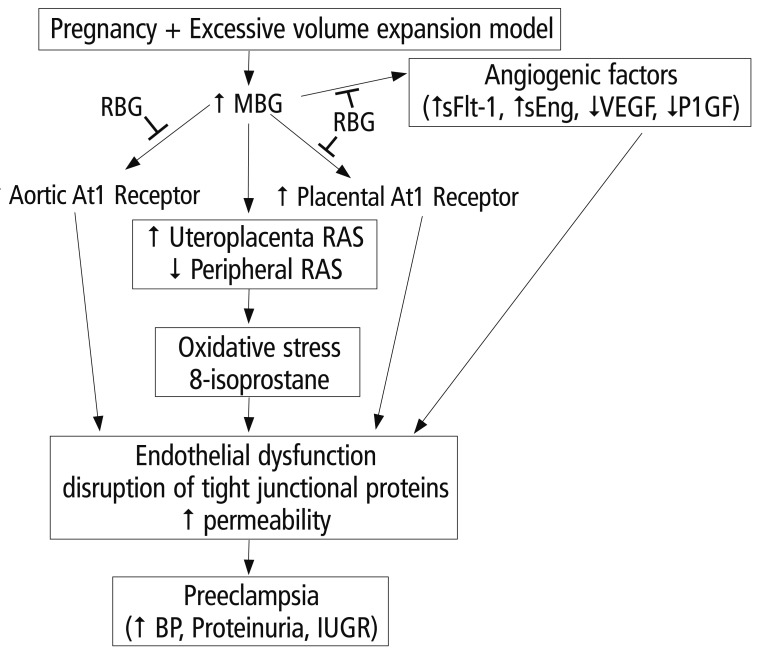
The renin-angiotensin system (RSA) is involved with regulating blood pressure. Circulating levels of angiotensin II increases as pregnancy advances [24]. In preeclampsia, circulating levels of angiotensin is decreased, despite increased vascular sensitivity to angiotensin with preeclampsia [25]. Uddin et al. [26] propose that MBG can act as a trigger of the RAS alterations observed in preeclampsia cases. Resibufogenin (RBG) is a recently discovered congener of MBG (Fig. 2B) that is postulated to act antagonistically to MBG [17]. Alterations in oxidative stress have been observed in the MBG rat model of preeclampsia [27]. MBG is involved in the alteration of oxidative stress, while RBG attenuates this alteration [17]. Excessive volume expansion in pregnancy causes an increase in the circulating levels of MBG that, in turn, causes an increase in the expression of RAS and the AT1 receptor. These increases produce oxidative stress and endothelial function. MBG also causes angiogenic imbalances, which ultimately lead to endothelial dysfunction (Fig. 3) [17].
Epigenetics and Micro-RNAs (miRNAs) in Preeclampsia
Recently, there has been a rapid development of new research in the field of epigenetics and fetal and maternal biology [28]. Major areas of epigenetic research include DNA methylation, histone modifications, and genomic imprinting. A promising new concept suggests the possibility that epigenetic dysregulations that occur prior to conception or during pregnancy might increase preeclampsia susceptibility [29]. Chelbi and Vaiman [30] suggest that abnormal methylation patterns may be an important mechanism underlying preeclampsia. In preeclampsia, cytotrophoblasts fail to deeply invade spiral uterine arteries. Shallow invasion of the spiral arteries leads to ischemic lesions and hypoxia. Chelbi and Vaiman [30] confirmed that SERPING1, SERPINE1, and SERPINE2-members of the serine protease inhibitor (SERPIN) gene family-are induced during a hypoxic state and show a modified expression pattern in placentas from preeclamptic pregnancies. Chelbi et al. [31] further found that the promotor of the serine protease inhibitor, SERPINA3, is hypomethylated in preeclamptic placentas compared with normal placentas. In particular, tissue inhibitor of metalloproteinase (TIMP3) showed the greatest degree of hypomethylation in early-onset preeclampsia placentas compared with control placentas, and TIMP3 is highly expressed in the preeclampsia placentas [32]. Increased TIMP3 expression may reduce the invasiveness of the trophoblasts during placental development, leading to placental hypoperfusion in early-onset preeclampsia [29].
Several studies have shown elevated plasma homocysteine concentrations in preeclampsia. Kulkarni et al. [33] found increased homocysteine and global DNA methylation levels in a group of preeclampsia cases compared with a control group. This study also found a positive association between global DNA methylation and systolic/diastolic blood pressure in the term preeclampsia group [33].
In addition to DNA methylation increases and histone modifications in the preeclamptic placentas, the expression patterns of miRNAs are very distinct. A recent study showed that miRNAs could play an important role in controlling DNA methylation and histone modification [34]. The differential expression of specific placental miRNAs (e.g., miR-210) was found to be upregulated in preeclampsia placentas compared with normal placentas [35].
Conclusion
To explain the pathogenesis of preeclampsia, there are several hypotheses including altered angiogenic balance, circulating angiogenic factors like marinobufagenin (a bufadienolide trigger), and activation of the renin-angiotensin system, among others. Epigenetically-modified circulating cell-free nucleic acids in plasma and serum could be novel markers with promising non-invasive clinical applications in the diagnosis of preeclampsia. We hope that these promising novel markers may eventually be able to reduce the morbidity and mortality of this severe disorder.
 XML Download
XML Download