

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bone regeneration requires the complex and well-organized process of bone formation.[1] In clinical fields, bone tissue regeneration has been a crucial part of regenerative medicine. For the realization of genuine bone regeneration from bench to bedside, tissue engineering techniques have been an important research topic.[2] Among the three key components of tissue-engineering (cells, scaffolds, and growth factors), cells cannot be artificially synthesized. Plentiful cell sources are necessary for the regenerative process; however, obtaining sufficient number of cells is challenging because of the limitation of the defect site. Therefore, various methods to obtain target cells have been investigated with the advances in stem cell research. To date, because of the limitations regarding the use of embryonic stem cells, the field of stem cell biology has expanded via the development of reprogramming technology using somatic cells.[3]
EPIGENETICS
Epigenetics is the study of changes in phenotype or gene expression that are somatically heritable but not caused by genetic alteration.[4] Epigenetic modifications including DNA methylation and histone acetylation could have long-term effects on gene expression.[5] Environmental factors such as nutrition, chemical compounds, stress, and other external factors could affect epigenetic modifications. This effect is apparent when environmental exposure occurs during gestation.[4] Epigenetic mechanisms that support transcriptional control of gene expression are important for the phenotypic diversity that emerges as a result of cellular differentiation during fetal development.[6] Chromatin is a complex of DNA, RNA, and protein in the nucleus.[7] DNA is packaged with histone proteins and forms the unit of the nucleosome like beads on a string structure. The chromatin structure is dependent on the transcription or replication state. Euchromatin is structurally loose and in an actively transcribed state or “turned on”, allowing for the binding of the transcription factor. On the other hand, heterochromatin is more condensed and is in an inactive state (“turned off”), blocking the approach of the transcription factor. Epigenetic chemical modification in chromatin also alters the structure, in particular DNA methylation and histone acetylation or methylation. 5′-cytosine methylation of 5′-CG-3′ dinucleotides (‘CpG doublets’) in genomic DNA is a prominent epigenetic modification that occurs at clusters of CpG doublets (‘CpG islands’) in transcriptional regulatory regions.[8] Generally, DNA hypermethylation of CpG islands in the promoter is related to chromatin condensation through repressive histone modifications that induce gene silencing.[9] DNA methyltransferases (DNMTs) and DNMT inhibitors regulate the methylation status of CpG islands after DNA replication in the cells in a specific manner.[10] The histone modification is a covalent post-translational modification (PTM) including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation. This PTM process alters the chromatin structure or recruits histone modifiers. Among them, histone acetylation and deacetylation are tightly involved with chromatin dynamics and transcription, gene silencing, and other cellular processes.[11] In most species, histone H3 is primarily acetylated at lysine residues 9, 14, 18, 23, and 56. H3 acetylation is usually increased by inhibition of histone deacetylases (HDACs) and decreased by histone acetyl transferases (HATs). Detecting which lysine of H3 is acetylated would provide useful information to better understand epigenetic regulation of gene activation. Likewise, both the state of DNA methylation and histone modification play essential roles in gene expression, and DNMT, HAT, and HDAC-related drugs could be good candidates for the treatment of target diseases.[1213] The mechanisms of environmentally induced phenotypes in humans remain unclear, making epigenetics research challenging.
THE CELL REPROGRAMMING METHODS WITH SOMATIC CELLS
1. Induced pluripotent stem cells (iPSCs)
iPSCs are pluripotent stem cells that are generated from somatic cells. Initially, the preparation of pluripotent cells was performed through the ectopic expression of master genes (Yamanaka factors), octamer-binding protein 4 (Oct4), sex-determining region Y-box 2 (Sox2), Kruppel-like factor 4 (KLF4), and avian myelocytomatosis viral oncogene homolog (c-Myc), in both embryonic and adult murine fibroblasts.[14] This remarkable achievement represented a big breakthrough in stem cell research. In the many studies using the Yamanaka factors, the potential for clinical application of iPSCs was established utilizing various mouse and human cell types.[1516] iPSCs have successfully been differentiated into a diverse range of cell and tissue types. Among them, the differentiation of iPSCs into mesenchymal cells or osteoprogenitor cells has actively been studied.[17] However, the possibility of iPSC de-differentiation into their original cell type was questioned because of the concept of epigenetic memory. The epigenetic memory of cells results from modifications to the cell's DNA that does not alter the DNA sequence, and it is inherited from the cell from which it descends. Such modifications can alter gene expression, inducing changes in the properties and behavior of the cells.
2. Trans-differentiation
Trans-differentiation, a process in which one somatic cell transforms into another somatic cell through bypass of the pluripotent state or progenitor cell condition, is well known as lineage switching or lineage conversion. Selman and Kafatos [18] first introduced the term ‘trans-differentiation’, based on the transformation of cuticle-producing cells to salt-secreting cells. Then, Eguchi and Okada [19] used the term to describe the phenomenon of conversion of chick retinal pigmented epithelial cells into lens fibers during the lens regeneration process. Recently, the concept of transdifferentiation has been re-evaluated taking into consideration the epigenetic stability of differentiated cells.[20] Previously, we reported the possibility of trans-differentiation from non-osteoblastic cells into osteoblastic cells through epigenetic modification.[6] Epigenetic modification with DNMTs inhibitor, 5-aza-2′-deoxycitidine (5-aza-dC) or HDAC inhibitor, trichostatin A (TSA), and osteogenic cue with bone morphogenetic protein 2 (Bmp2) or Wnt3a was applied to trans-differentiation. The 3T3-L1 pre-adipocytes [21] and human gingival fibroblast (HGF-1) gingival fibroblasts [22] were trans-differentiated into osteoblasts in vitro and induced new bone formation in vivo. These results indicated that epigenetic modification permits the direct programming of non-osteoblasts into functional osteoblasts, suggesting that this approach could be a novel therapeutic avenue in bone regeneration.
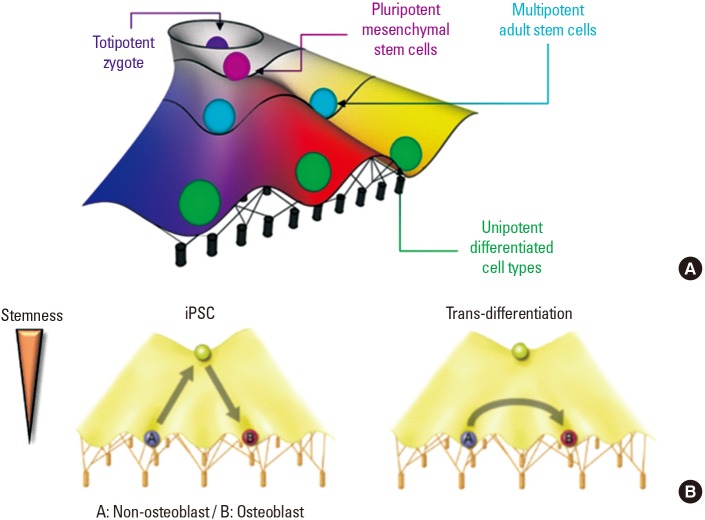
Waddington [23] suggested the concept of the ‘epigenetic landscape’, which is a metaphor for how gene regulation modulates cell development (Fig. 1A). In the epigenetic landscape metaphor, just as a marble rolls down a hill, stem cells lose their stemness and differentiate into somatic cells based on the surrounding environment. When we try to reprogram a cell from ‘A (ex. non-osteoblast)’ to ‘B (ex. osteoblast)’, in the case of iPSCs, the ‘A’ state should be de-differentiated into a stem cell and then re-differentiated into the ‘B’ state (Fig. 1B). However, trans-differentiation induces the jump from A to B, lowering the total time and effort through bypassing the induction of pluripotency. Likewise, direct trans-differentiation offers a more efficient route than the iPSCs reprogramming strategy. The latter requires initial regression of cells into a more immature state (de-differentiation) and subsequent induction of an alternative cell lineage. Hence, trans-differentiation has various advantages that could allow for its use in generating a next-generation cell source for tissue engineering.
TRANS-DIFFERENTIATION FROM NON-OSTEOBLAST CELLS TO OSTEOBLAST CELLS
Previously, we reported that Wnt3a stimulates Bmp2 expression, forming a positive autocrine loop in MC3T3-E1 pre-osteoblasts.[24] These cross-talk mechanisms are limited to osteogenic cells, which are stimulated to differentiate into mature osteoblasts in response to Bmp2 or Wnt3a (e.g., MC3T3-E1 pre-osteoblasts, C3H10T1/2 mesenchymal progenitor cells, ST2 bone marrow stromal cells, and C2C12 pre-myoblasts). Non-osteogenic cells that are finally differentiated into alternative mesenchymal cell lineages (e.g., 3T3-L1 pre-adipocytes and NIH3T3 fibroblasts) do not show Bmp2 expression in response to Wnt3a.[6] These differences between non-osteoblasts and osteoblasts indicate that an epigenetic mechanism may be related to this process.
It is well-known that (pre-) committed somatic cells do not typically change their phenotype, but there are some exceptions in which a (pre-) committed somatic cell is capable of transforming into another somatic cell type via trans-differentiation.[25] In a typical example, Bmp2 enables trans-differentiation from C2C12 pre-myoblasts into osteoblastic cells. In addition, natural trans-differentiation during newt lens regeneration provides insight into strategies for artificial trans-differentiation by direct reprogramming.[26] A number of mammalian programming strategies have recently emerged. For example, CCAAT/enhancer-binding protein (C/EBP) α and β efficiently convert differentiated B cells into macrophages,[27] Pancreatic duodenal homeobox-1 (PDX-1) drives trans-differentiation of adult hepatocytes into pancreatic cells,[28] and fibroblasts convert into cardiomyocytes using cardiac-related transcription factors and epigenetic remodeling proteins.[293031]
METHODS AND RESULTS OF TRANSDIFFERENTIATION
The 3T3-L1 pre-adipocytes,[21] NIH3T3 fibroblasts,[6] and HGF-1 gingival fibroblasts [22] were treated with 5′-aza-dC or TSA for 24 hr and then with Bmp2 or Wnt3a. Analysis of CpG islands of promoter lesions was performed using the University of California Santa Cruz (UCSC) genome browser software program. CpG islands exist in the target genes, which are related to osteogenesis and adipogenesis: Bmp2, alkaline phosphatase (ALP), runt-related transcription factor 2 (Runx2), and peroxisome proliferator activated receptor γ (PPAR-γ). Methylation-specific polymerase chain reaction (MSP) and chromatin immunoprecipitation (ChIP) assays were performed to analyze the methylation patterns. The DNA methylation states of CpG islands around the promoter were significantly different based on the cell characteristics. CpG islands of Bmp2, ALP, and Runx2 were highly methylated in the non-osteogenic cells; however, they were hypomethylated in the osteogenic cells. Based on the hypothesis that epigenetic differences between cells affect gene expression and determine the cell phenotype, we tried to modify the epigenetic background and then induce gene expression that is suppressed under typical conditions. 5′-aza-dC or TSA treatment in the non-osteoblast cells induced DNA hypomethylation or histone acetylation in the Bmp2, ALP, and Runx2 genes. This indicates that epigenetic modifications enable the transcription of genes inducing either the loose or active state of the chromatin structure. In accordance with epigenetic conditions, Bmp2, ALP, and Runx2 gene expression was increased by Bmp2 or Wnt3a treatment, despite the use of non-osteoblast cells. In vivo mouse studies also supported the possibility of trans-differentiation via epigenetic modification resulting in bone formation.
CONCLUSIONS
Our findings indicate that trans-differentiation of non-osteogenic cells into osteoblasts can be provoked by interfering with epigenetic inhibitory mechanisms. Based on these findings, we investigated bone regeneration through direct trans-differentiation from non-osteogenic cells to osteogenic cells by epigenetic modification. Epigenetic modification could permit the direct programming of non-osteoblasts into osteoblasts, and this approach might be a novel therapeutic avenue in bone regeneration.
 XML Download
XML Download