

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Humans have over 200 bones, which form the framework of the body. The biological functions of mammalian bones are numerous, including the support and movement of the body and production of blood cells. Bone is one of the important tissues for calcium homeostasis and storage. Furthermore, recent studies have uncovered a novel function of bone as an endocrine organ capable of secreting fibroblast growth factor 23 (FGF23) and osteocalcin.
Osseous tissues are formed through two distinct biological processes, intramembranous ossification and endochondral ossification.[1] During intramembranous ossification, osteoblasts produce type I collagen and bone-specific extracellular matrix (ECM) proteins, including osteocalcin, and then directly deposit calcium phosphate at the mineralization site. Thus, intramembranous ossification is induced by osteoblasts. In contrast, during endochondral ossification, cartilage anlagen are first generated through chondrogenesis and are eventually replaced by bone. Impaired endochondral ossification due to genetic mutation leads to various skeletal disorders, including achondroplasia. In addition to its role in physiological skeletal development, endochondral ossification is indispensable for fracture repair. Thus, understanding the molecular mechanisms underlying endochondral ossification is crucial for understanding the pathophysiology of genetic skeletal disorders and fracture healing.
In this review, we describe the process of endochondral ossification in detail and discuss the transcription factors critical at each stage, with a special focus on sex-determining region Y (SRY)-box 9 (Sox9) and runt-related transcription factor 2 (Runx2). We also review endoplasmic reticulum (ER) stress-related transcription factors, which have recently emerged as novel regulators of endochondral ossification.
MESENCHYMAL CONDENSATION
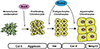
Endochondral ossification starts with the condensation of mesenchymal stem cells (Fig. 1). These cells are derived from the cranial and trunk neural crests, which produce the craniofacial mesenchyme and sclerotomes, respectively. Mesenchymal condensation is observed in E10.5 mouse limb buds and is characterized by densely packed mesenchymal cells. Although the precise molecular mechanisms are unclear, bone morphogenetic proteins (BMPs) are essential growth factors for mesenchymal condensation. The ectopic expression of Noggin, a BMP antagonist, in embryonic chick limb resulted in the complete absence of chondrogenesis.[2] Mouse genetic studies by Bandyopadhyay et al. [3] also confirmed that BMP is required for mesenchymal condensation. Using the Prx1 promoter, they deleted Bmp2 and Bmp4 before the onset of mesenchymal condensation and found that chondrogenic condensation did not occur in the mutant mice. In addition to BMPs, FGFs are necessary, as the deletion of FGF receptor (FGFR) 2 using Dermo1-Cre or Prx1-Cre impaired cartilage development.[45]
EARLY CHONDROGENESIS
In the formation of mesenchymal condensations, the cells at the center of the condensed area differentiate into chondrocytes and begin to produce chondrocyte-specific ECM components, including collagen type II α 1 chain (Col2a1) and aggrecan (Fig. 1). The abundant chondrocyte ECM components establish the cartilage primordia. These cells quickly proliferate and build up the chondrocyte layer observed in the growth plate. The proliferation of chondrocytes defines the shape of skeletal tissues by contributing to their directional elongation; most human bones are formed through this process. FGFs play an essential role in chondrocyte proliferation, and mutations of FGFR3 cause achondroplasia.[6]
1. Sox9
A large body of evidence has demonstrated that Sox9 is a critical transcription factor for chondrocyte differentiation from mesenchymal stem cells. It is a member of the SRY-related high-mobility group box family of proteins. The important role of Sox9 in skeletal development was first reported in studies of campomelic dysplasia, which is characterized by severe chondrodysplasia and autosomal XY sex-reversal. Two groups reported that mutations within and around Sox9 cause campomelic dysplasia.[78] In animal studies, Wright et al. [9] first reported the strong expression of Sox9 in embryonic mouse chondrocytes. Later, several groups studied the role of Sox9 in chondrogenesis and found that it directly regulates Col2a1 expression by binding to the chondrocyte-specific enhancer region located in the first intron of Col2a1.[1011,1213] A series of mouse genetic studies conducted by the de Crombrugghe group further demonstrated the essential role of Sox9 during early chondrogenesis. They generated Sox9-deficient chimeric mice and found that Sox9-null cells were excluded from mesenchymal condensation and that embryonic stem cells derived from Sox9(−/−) cells failed to form cartilage tissues.[14] They also found that embryonic cartilage development is defective in Sox9(+/−) mice, which died soon after birth due to cleft palate.[15] Akiyama et al. [16] reported that the deletion of Sox9 during mesenchymal condensation using Prx1-Cre completely prevents limb development. They also showed that chondrocyte-specific deletion of Sox9 in mice using Col2a1-Cre resulted in severe chondrodysplasia. Moreover, mutant mice in which Sox9 was deleted in cranial neural crest cells using Wnt1-Cre exhibited abnormal craniofacial development, including cleft palate.[17] In contrast, the forced expression of Sox9 during mesenchymal condensation using Prx1-Cre caused ectopic chondrogenesis characterized by polydactyly.[18]
2. Regulation of Sox9
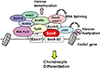
The regulatory mechanisms of Sox9 function during chondrocyte differentiation have been well-studied. The binding motif of Sox9 is CATTCAT in the Col2a1 enhancer [19] and CTCAAAG in the Col11a2 enhancer.[20] Sox9 regulates chondrocyte gene expression in collaboration with many transcriptional coactivators. Sox5 and Sox6, different classes of Sox family proteins, cooperatively induce the expression of Sox9 targets, including Col2a1 and aggrecan.[21] Double knockout mice for Sox5 and Sox6 showed severe chondrodysplasia.[22] Considering that Sox5 and Sox6 are not detected in chondrocyte-specific Sox9 knockout mice, Sox9 is necessary to induce Sox5 and Sox6.[16]
In addition to Sox5 and Sox6, several other transcription factors have been identified as transcriptional coactivators for Sox9. These coactivators are recruited to the transcriptional site of chondrocyte genes, and they directly or indirectly bind to Sox9 to form a large transcriptional complex of basic transcription factors (Fig. 2). Each component is involved in distinct modes of action in chondrocyte gene transcription. A functional gene screening system using the Col2a1 enhancer identified NonO/p54nrb, AT-rich interactive domain 5A (Arid5a), and zinc finger protein 219 (Znf219) as coactivators.[232425] NonO/p54nrb bound directly to Sox9 and regulated the mRNA splicing of Col2a1, suggesting that NonO/p54nrb links transcription and splicing with Sox9.[24] Our group also found that Arid5b recruits the histone demethylase Phf2 to Sox9 target genes and epigenetically regulates chondrogenesis.[26] Hattori et al. [27] identified Tat interactive protein-60 (Tip60) as a coactivator of Sox9 via yeast two-hybrid screening.[27] In addition to these molecules, β-catenin,[28] cAMP response element-binding protein (CREB)-binding protein (CBP)/p300,[29] thyroid hormone receptor-associated protein 230 (Trap230),[30] WW domain containing E3 ubiquitin protein ligase 2 (wwp2),[31] and PPARγ coactivator-1 (PGC-1α) [32] have been identified as Sox9-associated proteins.
Recently, two groups performed genome-wide chromatin immunoprecipitation-sequencing (ChIP-seq) studies to uncover the precise regulatory mechanism of Sox9 function. Liu and Lefebvre [33] performed ChIP-seq analysis using anti-Sox9 and found that Sox9 recognizes pairs of inverted Sox motifs and binds to most super-enhancers present in chondrocytes. They identified FGFR3, Runx2, and Runx3 as target genes of the Sox trio. Ohba et al. [34] also performed ChIP-seq analysis, using a variety of antibodies against Sox9 and the epigenetic markers of histone modification. They analyzed the epigenetic status of Sox9 target genes in detail and proposed two distinct modes of Sox9 function in chondrocytes.[34] In class I engagement, or “indirect binding via the basal transcriptional complex,” Sox9 recognizes the transcriptional start sites of non-chondrocyte-specific genes. In class II engagement, or “binding to multiple active enhancer elements through sub-optimal, low-affinity Sox dimeric motifs,” Sox9 forms dimers and binds to multiple super-enhancer regions of chondrocyte genes. These studies are crucial to understanding the precise mechanism of Sox9 function during chondrogenesis.
ER STRESS-RELATED TRANSCRIPTION FACTORS
The ER is susceptible to various stresses that provoke the accumulation of unfolded proteins in the ER lumen. Eukaryotic cells deal with unfolded proteins accumulating in the ER with the unfolded protein response. Chondrocytes need to produce a large amount of ECM components in the ER over a short period to generate a cartilage template; therefore, physiological ER stress continuously occurs in early chondrocytes. Recent reports have uncovered the novel function of ER stress in skeletal development through ER stress-related transcription factors.
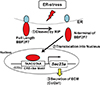
BBF2 human homolog on chromosome 7 (BBF2H7) is a major transmembrane transcription factor with a basic leucine zipper domain, which belongs to the CREB/activating transcription factor (ATF) family. It is preferentially expressed in chondrocytes, and the full-length protein localizes to the ER. In response to ER stress, BBF2H7 is cleaved at the membrane by regulated intramembrane proteolysis, dividing it into two molecules (Fig. 3). The N-terminal fragment translocates into the nucleus, where it acts as a transcription factor to activate its target gene Sec23 homolog A, coat complex II component (Sec23a) (Fig. 3). Sec23a is a COPII vesicle component that is essential for ER–Golgi transport; therefore, Sec23a plays an essential role in the secretion of chondrocyte-specific ECM components, including Col2a1. The deletion of Bbf2h7 causes severe chondrodysplasia due to the abnormal accumulation of type II collagen in the ER.[35]
Atf4 is also a member of the CREB/ATF family with a leucine zipper domain. The translation of Atf4 is controlled by the phosphorylation of eukaryotic initiation factor 2α, which is mediated by PKR-like ER kinase in response to ER stress. Atf4 regulates chondrocyte proliferation and differentiation by activating Indian hedgehog (Ihh) transcription in chondrocytes.[36]
CHONDROCYTE HYPERTROPHY
During endochondral ossification, chondrocytes stop proliferating and become prehypertrophic chondrocytes, which express specific markers, including Ihh and parathyroid hormone (PTH)/PTH-related protein (PTHrP) receptor. Then, prehypertrophic chondrocytes enlarge and become hypertrophic chondrocytes. Prehypertrophic chondrocyte cell volume increases as much as 10- to 15-fold over proliferating chondrocytes, and cellular hypertrophy occurs mainly via cell swelling by water imbibition.[37] Cooper et al. [38] conducted a detailed analysis of chondrocyte hypertrophy and found that it involves three phases, one of which is dependent on insulin-like growth factor. Hypertrophic chondrocytes undergo terminal differentiation and produce matrix metalloproteinase 13 (MMP13) to remodel the ECM, which allows vascular invasion followed by bone formation. Hypertrophic chondrocytes eventually die by apoptosis. Although the mechanism is not fully understood, several transcription factors, including Runx2, myocyte enhancer factor 2c (Mef2C), and forkhead box (Fox) proteins, have been reported to regulate chondrocyte hypertrophy.
1. Runx2
Runx2 was first identified as a master transcription factor driving osteoblast differentiation by searching for molecules that bind directly to the osteoblast-specific enhancer region located in the osteocalcin gene promoter.[39] The essential role of Runx2 in chondrocyte hypertrophy was studied by gain of function and loss of function approach of mutant mouse. Takeda et al. [40] reported that forced expression of Runx2 using Col2a1-Cre induced ectopic Col10a1 expression in columnar chondrocytes. The Komori group at Osaka University found that terminal differentiation of chondrocytes was delayed in Runx2 knockout mice, [41] and later, demonstrated that hypertrophic chondrocytes were completely absent in Runx2(−/−)3(−/−) mice.[42] Runx2 directly transcribes Ihh,[42] Col10a1,[43] and Mmp13 [44] in chondrocytes, and it controls these genes in collaboration with several transcription factors, including CCAAT/enhancer-binding protein β (C/EBPβ). Because MMP13 contributes to the cartilage destruction during the development of osteoarthritis, Runx2 can be the molecular target for the treatment of osteoarthritis.
2. Mef2c
Mef2c, which is involved in muscle development, is expressed in hypertrophic chondrocytes, and its deletion in mice impairs chondrocyte hypertrophy.[45] There are several Mef2-binding sites in the Col10a1 gene promoter and Mef2c activates Col10a1 gene promoter activity through direct binding to these elements. It has also been demonstrated that deletion of one Mef2c allele in Hdac4(−/−) mice partially rescues the abnormal ossification observed in Mef2c(−/−) mice. Interestingly, Verzi et al. [46] generated neural crest-specific Mef2c knockout mice using Wnt1-Cre, and the mice lacking Mef2c exhibited severe defects in craniofacial development. These reports collectively suggest that the balance between transcriptional activation by Mef2c and inhibition by Hdac4 is critical not only for chondrocyte hypertrophy but also for skeletal development.
3. Fox transcription factors
Fox proteins comprise a large family of transcription factors. Among over 50 Fox genes, FoxA2/A3 and FoxC1/C2 regulate chondrocyte hypertrophy through the direct induction of Col10a1. Accordingly, chondrocyte hypertrophy was delayed in Col2–Cre, FoxA2flox/flox, and FoxA3−/− mice.[47] Our group identified FoxC1 as a novel chondrogenic transcription factor through fluorescence-activated cell sorting-assisted microarray analysis using Col2a1-Venus mice in which chondrocytes are selectively labeled with fluorescent proteins. FoxC1 increased Col10a1 expression via direct binding to the Col10a1 gene promoter, and spontaneous Foxc1 mutant mice, called Ch mice, were completely devoid of Col10a1-positive hypertrophic chondrocytes during sternal development.[48] In addition to Col10a1, the overexpression of FoxC1 in primary chondrocytes promoted the expression of PTHrP, a target gene of Ihh-Gli2 signaling in chondrocytes. FoxC1 physically associates with Gli2 and increases the DNA binding of Gli2 to Ihh target gene promoter. This functional interaction leads to the elevated expression of Gli1 and protein patched homolog 1 (Ptch1), another target gene of Ihh in chondrocytes. Interestingly, the pathogenic missense mutation of FOXC1 (F112S), which causes Axenfeld–Rieger malformations, failed to interact with Gli2 and decreased PTHrP expression. Our findings collectively suggest that the functional interaction between FoxC1 and Ihh-Gli2 signaling is critical for endochondral bone formation and the impaired functional interaction caused by pathogenic mutation contributes to the pathogenesis of Axenfeld–Rieger syndrome.
PROSPECTS
In this review, we introduced a small number of the transcription factors that control endochondral ossification. However, endochondral ossification is complicated and highly organized, and it may involve as yet unknown transcription factors. Moreover, the transcriptional activities of these chondrogenic transcription factors are modulated through posttranscriptional modification. Therefore, the molecular mechanisms underlying endochondral ossification have not been fully elucidated. The molecular mechanism by which Sox9 is induced in mesenchymal condensation is the major question that should be addressed. Fortunately, recent studies have revealed novel mechanisms underlying endochondral ossification. For instance, several groups reported an unexpected function of Sox9 in promoting chondrocyte hypertrophy using ChIP-seq analysis [49] and a sophisticated mouse genetic approach.[50] We believe that the further analysis of endochondral ossification will uncover the novel molecular mechanisms of skeletal development and that these basic research findings will contribute to the development of treatments and therapies for skeletal abnormalities.
 XML Download
XML Download