

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ROLE OF Sox9 IN CARTILAGE DEVELOPMENT
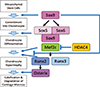
In mammals, cartilage predominantly forms via endochondral ossification, which is regulated by several transcription factors.[12] Genetic studies clearly evident that sex-determining region Y (SRY)-box 9 (Sox9) plays an essential role in the initiation stage of cartilage development (Fig. 1). Cartilage-specific Sox9 conditional-deficient mice fail to form cartilage.[3] Moreover, mutations of the human SOX9 gene cause campomelic dysplasia, which manifests abnormalities in cartilage formation.[45] Sox9 has been identified as a transcription factor that binds to the critical cis element present in the collagen type II α 1 chain (Col2a1) gene.[6] Sox9 stimulates differentiation of mesenchymal cells into chondrocytes and upregulates early chondrogenic genes including Col2a1, Col11a2, and aggrecan.[7] Although direct binding of Sox9 to Sox5 and Sox6 remains unclear, Sox5 and Sox6 have been shown to be essential transcriptional partners of Sox9 during cartilage development (Fig. 1). Indeed, Sox5 and Sox6 double knockout mice display severe cartilage defects.[8] Overexpression of Sox5 and Sox6 markedly enhances the chondrogenic action of Sox9.[79] Importantly, expression of Sox5 and Sox6 are not observed in cartilage-specific Sox9 conditional-deficient mice,[3] indicating that Sox5 and Sox6 function downstream of Sox9. To support this, overexpression of Sox9 has been shown to upregulate Sox5 and Sox6 expression.[7] Thus, Sox9, Sox5, and Sox6 function as essential transcription factors in the early stage of cartilage development.
In contrast to the role of Sox9 during early-stage cartilage development, Sox9 seems to negatively regulate the late stage of endochondral ossification (Fig. 1). Misexpression of Sox9 in murine hypertrophic chondrocytes markedly suppresses vascular invasion and calcification during endochondral ossification.[10] Consistently, expression of vascular endothelial growth factor (VEGF) that plays a central role in vascular invasion was diminished in the mice.[10] Conversely, Sox9 interacts with Indian hedgehog (Ihh)/Gli2 signaling,[11] therefore stimulating the effect of Ihh/Gli2 on induction of parathyroid hormone-related protein (PTHrP) strongly inhibits the late stage of endochondral ossification.[11] Recently, forkhead box C1 (Foxc1) has been demonstrated to upregulate PTHrP expression in cooperation with Ihh/Gli2 signaling (Fig. 2).[12] Although the relationship between Sox9 and Foxc1 remains to be elucidated, it is possible that Sox9, Foxc1 and Gli2 cooperatively control endochondral ossification by regulating PTHrP expression. Sox9 likely forms a negative-feedback loop for the late stage of endochondral ossification through regulation of VEGF and PTHrP expression (Fig. 2).
REGULATION OF Sox9 DURING CARTILAGE DEVELOPMENT
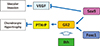
In addition to Sox5 and Sox6, several other transcription factors and transcriptional regulators that interact with Sox9 and regulate its function during cartilage development have been identified (Fig. 3). PPARγ coactivator-1α (PGC1α) has been identified as a transcription factor specifically expressed in developing limb buds and shown to interact with Sox9 to stimulate cartilage development.[13] One study using knock-in mice, into which the green fluorescent protein (GFP) gene was introduced into the Sox9 gene, revealed involvement of the WW domain containing E3 ubiquitin protein ligase 2 (Wwp2) in cartilage development (Fig. 3).[14] Wwp2 also interacts with Sox9 and mediator complex subunit 25 (Med25) to regulate cartilage development.[14] Another study indicates that Wwp2 controls cartilage development through monoubiquitination of goosecoid, which regulates Sox6 expression.[15] AT-rich interactive domain 5A (Arid5a), Zfnf219 and p54nrb, isolated using a mammalian cell expression cloning system,[16] have been shown to interact physically and functionally with Sox9 (Fig. 3).[71718] Arid5a appears to stimulate cartilage development, presumably by controlling chromatin remodeling of chondrogenic genes.[17] Because Arid5a is involved in interleukin-6 mRNA stability and inflammation, [19] it is possible that this transcription factor might be associated with cartilage diseases such as rheumatoid arthritis. Zinc finger protein 219 (Znf219), which is specifically expressed in limb buds, is also implicated in chondrocyte differentiation.[7] p54nrb conducts splicing of chondrogenic genes in cooperation with Sox9.[18] p54nrb is a known regulator of the newly-proposed paraspeckle bodies.[18] Determining how the paraspeckle body is regulated during cartilage development is of strong interest. RNA-Seq analysis indicates that Arid5b is highly expressed in C3H10T1/2 cells that have the ability to differentiate into chondrocytes, but not in NIH3T3 fibroblasts.[20] Arid5b interacts with a histone demethylase, PHD finger protein 2 (Phf2), and recruits Phf2 to promoter regions of chondrogenic genes, subsequently stimulating the chondrogenic action of Sox9.[20]
Regulatory mechanisms of Sox9 expression in mesenchymal cells remain elusive, although transient receptor potential cation channel subfamily V member 4 (TRPV4) is implicated in Sox9 expression.[16] Specific transcription factors necessary for induction of Sox9 are yet to be identified. To address this, combination studies using bioinformatics and updated molecular cloning approaches in mesenchymal cells might be effective and powerful. In addition, analyses of downstream TRPV4 signaling in the nucleus should be addressed. These studies will advance our understanding of Sox9 regulation during cartilage development.
REGULATION OF THE LATE STAGE OF ENDOCHONDRAL OSSIFICATION BY TRANSCRIPTION FACTORS
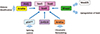
Chondrocyte hypertrophy is a key biological event during endochondral ossification, during which both cell morphology and gene expression profile change dramatically. It is clear that Runx2 and Runx3 play an essential role in chondrocyte hypertrophy because hypertrophic chondrocytes are absent in Runx2 and Runx3 double knockout mice (Fig. 1).[21] Since Runx2-deficient mice show severe impairment of chondrocyte hypertrophy,[21] Runx2 seems to be the dominant regulator in the step. Furthermore, Runx2 regulates Ihh expression in chondrocytes.[21] Furthermore, Ihh stimulates chondrocyte maturation in a PTHrP-independent-manner [22] and inhibits chondrocyte hypertrophy through upregulation of PTHrP expression. Because interaction between Runx2 and Ihh/Gli2 signaling has been shown,[23] Runx2 appears to regulate chondrocyte maturation by forming a synergistic loop with Ihh/Gli2 signaling (Fig. 4). As expected, the necessity of core binding factor β (Cbfβ), a well-known co-activator for Runx family members, in cartilage development is clearly demonstrated in vivo (Fig. 4).[24] CCAAT/enhancer-binding protein β (C/EBPβ) and activating transcription factor 4 (ATF4) associate with Runx2 and regulate osteoblast differentiation (Fig. 4).[2526] Similarly, C/EBPβ and ATF4 play an important role in cartilage development, especially in the late stage,[2728] indicating that both C/EBPβ and ATF4 function as transcriptional partners for Runx2 during endochondral ossification (Fig. 4).
Myocyte-enhancer factor 2C (Mef2C) is expressed in hypertrophic chondrocytes and is indicated to be critical for early-stage hypertrophy of chondrocytes (Fig. 1).[29] Of note, cartilage-specific Mef2c-deficient mice show impaired expression of Col10a1 and Runx2.[29] In vitro analyses demonstrate direct regulation of Col10a1 expression by Mef2c.[29] Similarly, Mef2c seems to regulate Runx2 expression through a specific novel cis element with distal-less homeobox 5.[30] Interestingly, phenotypes of heterozygous Mef2c mutant mice are rescued by crossing with histone deacetylase 4 (Hdac4) null mice, suggesting an antagonistic effect of Hdac4 on Mef2c during endochondral ossification (Fig. 1).
In vitro studies indicate that hypoxia-inducible factor-2α (HIF2α) stimulates expression of Col10a1, matrix metalloproteinase 13 (MMP13), and VEGF.[3132] Consistent with this, heterozygous HIF2α mutant mice show suppression of chondrocyte hypertrophy and reduction in Col10a1, MMP-13 and VEGF expression.[3132] Taking these findings together, HIF2α plays an important role in cartilage development. In addition, RELA proto-oncogene (RelA), a component of nuclear factor-κB (NF-κB), seems to function as an inducer of HIF2α in chondrocytes.[31]
Osterix is a transcription factor specifically expressed in prehypertrophic chondrocytes. Studies in Osterix knockout mice indicate that this transcription factor is required for calcification and degradation of cartilage matrices.[33] Furthermore, Osterix null mice fail to form cartilage matrix vesicles.[33] Interestingly, chondrocyte hypertrophy is almost intact in Osterix-deficient mice.[33] Moreover, Osterix directly regulates MMP13 expression through physical association with Runx2.[33] Consequently, it is likely that Osterix plays a critical role in the final stage of cartilage development (Fig. 1).
OSTEOARTHRITIS AND TRANSCRIPTION FACTORS
Osteoarthritis is one of the most common age-related cartilage diseases. Because several of the morphological and gene expressional changes seen in osteoarthritis patients resemble events in the late stage of endochondral ossification, particularly the hypertrophy stage, several investigators have attempted to understand pathogenic roles of the transcription factors associated with late-stage endochondral ossification in osteoarthritis.
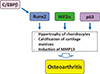
Runx2 is thought to be associated with the pathogenesis of osteoarthritis, since heterozygous Runx2-deficient mice are less affected by knee joint instability in comparison with wild-type mice (Fig. 5).[34] However, involvement of Runx2 in osteoarthritis is not reported in cleidocranial dysplasia patients. Haploinsufficiency of the Runx2 gene may therefore not be critical for osteoarthritis pathogenesis.
HIF2α appears to be associated with the pathogenesis of osteoarthritis (Fig. 5). Heterozygous HIF2α-deficient mice are resistant to treatments that cause osteoarthritis.[3132] Moreover, association of single nucleotide polymorphism (SNP) of the HIF2α gene with osteoarthritis is reported.[31] In contrast, replication of HIF2α SNP in knee osteoarthritis is absent.[35] Cartilage-specific HIF2α-deficient mice also show very modest impairment of endochondral ossification.[36] Thus, further dissection of the pathogenic role of HIF2α in osteoarthritis is necessary.
The possible mechanisms by which C/EBPβ is implicated in the pathogenesis of osteoarthritis are shown (Fig. 5). MMP3, MMP13 and a disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS-5) mRNA levels are upregulated following overexpression of C/EBPβ.[37] In addition, C/EBPβ is associated with inflammatory arthritis through mediating MMP-13 expression.[38] More interestingly, cooperation of C/EBPβ and Runx2 in osteoarthritis has been shown in vivo, and HIF2α appears to function upstream of C/EBPβ.[27]
More recently, the importance of p63, a family member of p53, has been indicated in osteoarthritis (Fig. 5).[39] Cartilage-specific p63-deficient mice are resistant to the development of osteoarthritis by inhibition of chondrocyte apoptosis. Notch signaling is also involved in the pathogenesis of osteoarthritis by controlling Col10a1, MMP13 and VEGF expression.[40] Consistently, hairy and enhancer of split 1 (Hes1), a target transcription factor of Notch signaling, has been shown to be associated with osteoarthritis.[41] Similarly, new mechanisms of osteoarthritis development have been proposed using mouse genetic studies.
ARTICULAR CHONDROCYTES AND OSTEOARTHRITIS
As described above, the identification of transcription factors involved in the pathogenesis of osteoarthritis is progressing rapidly. These studies will certainly contribute to developing more effective and novel therapies for osteoarthritis. However, considering that articular chondrocytes have distinct cellular and molecular properties from growth plate chondrocytes, further investigations using articular chondrocytes will be necessary to better understand osteoarthritis pathogenesis and to develop more effective therapies for the disease. One of the most straightforward therapies for osteoarthritis is regeneration of cartilage in the damaged joints. Direct reprogramming of skin fibroblasts into chondrocytes is reported.[42] Although this technology has the advantage to negate the issues surrounding induced pluripotent stem cells, the regenerated cartilage appears to calcify. Another study reports the effect of growth differentiation factor 5 (GDF5) on cartilage regeneration without calcification of the cartilage tissues.[43] Indeed, GDF5 is a well-known marker specific for articular cartilage.[44] It is therefore important to identify transcription factors critical for GDF5 regulation. Such investigation might further advance cartilage regeneration technology. Importantly, several microRNA, including miR-140, miR-145, and miR-675, are involved in pathogenesis of osteoarthritis by regulating transcription and/or translation of chondrogenic genes.[45] It is, therefore, likely that these microRNA would be good therapeutic targets for osteoarthritis.
Because the molecular mechanisms of articular cartilage destruction in osteoarthritis patients are also elusive, investigation focused on articular chondrocytes would provide novel insight into better understanding of osteoarthritis pathogenesis. Moreover, gene expression profiling of articular chondrocytes could be helpful to understanding cellular and molecular properties of articular chondrocytes.

 XML Download
XML Download