

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Wilson's disease (WD) is an autosomal recessive disorder of biliary copper excretion and is characterized clinically by hepatic, neurological and ophthalmic manifestations related to the accumulation of copper in the liver, the lenticular nuclei of brain and cornea respectively. WD is found worldwide, however, it has higher prevalence in some isolated populations due to high rates of consanguinity.[1]
However, due to social stigma attached to patients of WD and a lack of thorough work up of suspected cases, WD is under-reported. Furthermore, when WD is diagnosed, medical care is focused on the neurological and hepatic manifestations. This has led to under-recognition of orthopaedic manifestations of WD. Here we present a case of pathological fracture of neck of femur in child who was subsequently diagnosed with WD. A written informed consent was obtained from the parents of the patient authorizing radiological examination, photographic documentation, and surgery. They were also informed that the data concerning the case would be submitted for publication and they consented.
CASE
A 12-year-old girl, weighing 25 kg, was brought by her parents to our out patients department with complaints of inability to bear weight on the right lower limb since 3 months. There was no history of significant trauma. Her parents revealed a history of normal infancy and early childhood followed by delayed milestones from 3 years of age onwards, mental stunting, loss of ability to speak proper words, drooling, and abnormal involuntary movements of upper limbs. The child was a product of a consanguineous marriage and had two younger siblings, who were apparently free of inherited diseases. The parents in the past had consulted various pediatricians during the patients early childhood, but reported that no conclusive diagnosis had been made. Prior to the development of current complaints, she was able to walk independently with a jerky, clumsy gait as described by the parents.
General physical examination revealed short stature, thin build, mask–like facies, hypertonia and flexion attitude of elbows, knees and hips. She had abnormal choreoathetoid movements of both upper limbs on rest. The right hip was tender on palpation and patient did not allow assessment of passive movements due to pain. The limb was kept in an attitude of adduction, flexion of 70° at hip and 120° at knee. The greater trochanter was more prominent on palpation on the affected side, but patient did not allow assessment of telescopy and active straight leg raising. Any attempted movement of the knee was also resisted by the patient, which had led to development of flexion contracture at hip and knee. Our provisional diagnosis was a non-union of pathological fracture of neck of femur.
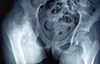
Radiographic examination of the hip revealed a suspected varus malunion of fracture neck of femur on antero-posterior view (Fig. 1). However, to confirm the diagnosis, lateral view radiograph of the hip could not be obtained due to inability to place the patient in the desired position. Computed tomography was considered, but could not be done due voluntary and involuntary movements of the patient. A clinical and fluoroscopic examination under anesthesia was planned.
Biochemical investigations revealed mostly normal parameters except for mildly deranged liver functions. A pediatric consultation was sought to look for a syndromic diagnosis. Based on their suspicion of WD, and ophthalmic examination was arranged. Slit lamp examination confirmed the presence of Kayser-Fleischer rings in the limbus of the cornea. Further laboratory investigations revealed a serum ceruloplasmin of 8.8 mg/dL (normal range, 20–50 mg/dL), free serum copper of 54 µg/dL (normal range, 10–15 µg/dL) and 24 hr urinary copper excretion of 137 µg/24 hr (normal range, 10–30 µg/24 hr), thus confirming the diagnosis of WD.
Patient was started on oral copper chelator D-penicillamine. This was supplemented by oral pyridoxine. To control the neurological symptoms, patient was also started on oral trihexyphenidyl.
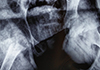
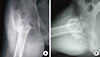
One week after starting these medications, the patient was taken for clinical and fluoroscopic examination under general anaesthesia. The examination revealed telescopy at fracture site, positive Desault sign and mobility at fracture site was noted under fluoroscopy on push and pull stress. Hence a diagnosis of non-union of fracture of neck of femur was confirmed. Considering the poor quality of bone and hence poor purchase that the bone would afford to implants, a classical McMurray's osteotomy (oblique intertrochanteric lateral closing wedge valgus osteotomy with medial displacement of proximal end of distal fragment) was done along with adductor tenotomy (Fig. 2). The fracture was immobilized in a one and half hip spica with operated hip in 30° abduction. Post-operative period was uneventful. Zinc acetate was added along with oral calcium supplementation and weekly cholecalciferol in granule form. At four weeks, post-operatively, the spica cast was modified by bringing the involved hip to neutral in coronal plane whilst taking the opposite hip to 45° abduction. McMurray [2] in his description of the osteotomy in adult patients removed the spica at three and a half months. However, since our patient was a child, expecting an early union, we removed the spica at three months post-operatively for clinical and radiographic assessment. Patient had no pain or tenderness and allowed passive abduction from neutral to 30°. Passive flexion was permitted from 10° to 90°. She was able to perform actively raise her leg off the bed. Antero-posterior and lateral views revealed signs of union at fracture site (Fig. 3). For 4 weeks, lower limb strengthening exercises were done to regain the strength lost during spica immobilization. At 4 months, follow up patient was able to ambulate with support. At 6 months follow up, an unsuccessful attempt was made to ambulate the patient without support, however she continued to ambulate within the household with support.
DISCUSSION
Wilson [3] first described this disease as a distinct clinical entity in 1912, when he reported four patients with this familial disease. Since then many researchers have made significant contributions to our understanding of the disease (Table 1).
WD is an autosomal recessive inherited disorder with an estimated prevalence of 1 case per 30,000 live births. Most studies claim equal prevalence in men and women, however the largest registry of WD patients shows a slight male predominance.[456]
The abnormal gene for WD is localized to the long arm of chromosome 13 (13q14.3). This gene encodes a copper transporting P-type adenosine triphosphatase (ATPase), ATP7B, which is mainly but not exclusively expressed in hepatocytes.[7] It is critical for biliary copper excretion and copper incorporation into ceruloplasmin. More than 200 mutations have been described in the ATP7B gene on chromosome 13 in patients with WD. Absence or malfunction of ATP7B gene results in decreased biliary copper excretion and diffuse accumulation of copper in the cytosol of hepatocytes. With time, liver cells become overloaded and copper is redistributed to the brain, kidneys and eyes. Ionic copper inhibits pyruvic oxidase in brain and ATPase in membranes.[8]
Mutations that completely knock out gene function are associated with an early onset of symptoms at 2 to 3 years of age, when WD might not typically be considered in the differential diagnosis. Although missense mutations are most frequent, deletions, insertions, nonsense, and splice site mutations all occur. Individuals with the same mutation, even homozygotic twins, may demonstrate wide variability in age of symptom onset and clinical presentation.[7]
The basic pathogenic defect lies in the liver, which makes hepatic manifestations the most frequent presentation. WD may present as hepatitis, cirrhosis, or as hepatic failure, typically in the mid to late teenage years. Hemolytic anemia may occur because of large amounts of copper released by hepatocellular necrosis. The neurologic manifestations typically occur due to damage in the basal ganglia giving rise to dystonia, incoordination, and tremor. These are associated with psychiatric disturbances like emotional lability or depression. Cholelithiasis, nephrolithiasis, hematuria, sunflower cataracts and Kayser-Fleischer rings (copper deposits in the outer rim of the cornea) may also be seen.
Hepatic and neuropsychiatric manifestations along with Kayser-Fleischer rings seen under slit lamp are usually diagnostic. The biochemical diagnosis of WD is based on decreased serum ceruloplasmin, increased free serum copper, decreased total serum copper (due to decrease of ceruloplasmin), increased hepatic copper content, and increased urinary excretion of copper. Genetic testing may be helpful for detection of specific mutation and counseling.
Due to a myriad variety of clinical features there is tendency for the less obvious musculoskeletal features to go unnoticed. Relatively well-known musculoskeletal manifestations of WD include synovitis,[9] early osteoarthritis,[9] osteoporosis,[10] rickets and osteomalacia.[11] Lesser known manifestations include spontaneous fracture,[12] heterotopic ossification,[13] scoliosis,[14] epiphyseal dysplasia,[14] genu varum,[15] avascular necrosis of femoral head [15] and osteochondritis dissecans.[16]
Recently, the musculoskeletal aspect of WD has begun to receive its due importance. In 2009, a new Global assessment scale for WD was launched, which included osseomuscular involvement as one of the four key domains.[17] Yu et al. [15] in 2017, described this osseomuscular type of WD, which lacks the characteristic hepatic and neurological features and hence has a tendency to be misdiagnosed.
In spite of these recent developments, after a thorough literature search, we could find only two reported cases of fractures leading to the diagnosis of WD. Shin et al. [18] reported a case of a 25-year-old male who presented with fracture of radius and ulna following trivial trauma. The patient already had subtle neurological signs of WD, however it was diagnosed while investigating the cause of osteoporosis in the patient. Verma et al. [12] reported a case of a 16-years-old male presenting with pathological fracture of the humerus along with full-blown hepatic, neuropsychiatric and ophthalmic features of WD.
The main stay of treatment of fractures in WD remains the same as for any other fracture. However, prevention of fractures and morbidity due to skeletal changes should be the prime focus. This requires vitamin D supplementation and replacement of calcium and phosphorus, which are lost in urine. Copper accumulation is treated by chelation therapy with D-penicillamine, trientine and/or zinc. Diet should also be modified to minimize copper intake by avoiding chocolate, liver, nuts, mushrooms and avoiding the use of copper utensils for cooking.
Liver transplantation should be considered in patients with decompensated liver disease.
CONCLUSION
WD is an uncommon but important cause of pathological fractures in the young population and must be considered in the differential diagnosis of metabolic bone disorders. A high index of suspicion should be maintained for patients presenting with hepatic or neurological disease along with poor bone quality or pathological fractures.

 XML Download
XML Download