

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Osteoclasts are terminally differentiated multinucleated cells responsible for resorbing bone matrix and are also critical to the maintenance of bone remodeling and mineral homeostasis.[12] These cells are generated by the proliferation, fusion, and differentiation of hematopoietic precursors of the monocyte/macrophage lineage. Two molecules, receptor activator of nuclear factor-kappa B (NF-kB) ligand (RANKL) and macrophage colony-stimulating factor (M-CSF) are essential and sufficient for osteoclast development.
M-CSF initiates cellular responses by binding to its receptor, c-Fms. Binding of M-CSF triggers receptor dimerization and tyrosine autophosphorylation, which, in turn, leads to the activation of downstream signaling pathways such as mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) and phosphatidylinositol (PI)3/Akt. These cellular events provide the signals necessary for proliferation and survival of osteoclast precursor cells. While the mitogen-activated protein kinase (MAPK) pathway, MEK/ERK, primarily supports proliferation of macrophages (osteoclast precursors),[345] PI3K/Akt mediates survival,[6] proliferation,[7] and cytoskeletal organization.[8]
Interaction of RANKL with its receptor, RANK, stimulates the induction and activation of several transcription factors including NF-κB, c-Fos, and nuclear factor of activated T cells c1 (NFATc1), all of which are crucial for osteoclast differentiation. NFATc1 can then induce the expression of osteoclastogenic genes such as tartrate-resistant acid phosphatase (TRAP) and cathepsin K in cooperation with other transcription factors including AP-1 and PU.1.[9]
Lipocalin-2 (LCN2), also known as neutrophil gelatinase-associated lipocalin (NGAL), is a small glycoprotein belonging to the lipocalin family, which functions as a carrier, transporting lipids and other hydrophobic molecules under physiological conditions. LCN2 was initially reported to exert antibacterial activity by chelating bacterial siderophores.[1011] Consequently, LCN2-deficient mice are highly sensitive to bacterial infection.[12] Moreover, LCN2 plays an important role in various cellular processes, such as cellular survival,[131415] proliferation,[10111617] and differentiation.[1819]
We recently demonstrated that LCN2 and its receptors, megalin and 24p3 receptor (24p3R), are expressed in osteoclast lineage cells.[20] Additionally, we demonstrated that ectopic expression or treatment of recombinant LCN2 suppresses proliferation and differentiation of osteoclast lineage cells, thus inhibiting osteoclast formation.[20] In the present study, we further investigated the role of LCN2 in osteoclast development mediated by M-CSF and RANKL, using a LCN2-deficient mouse model (LCN2-/- mice). Results showed that LCN2 deficiency enhances macrophage proliferation and osteoclast differentiation in vitro; events that are related to increased c-Fms expression and RANKL-induced NF-κB activation. Hence, these observations led us to examine bone phenotype of LCN2-/- mice, to determine the role of LCN2 in vivo.
METHODS
1. Mice and analysis of bone phenotype
LCN2-/- mice were provided by Dr. Shizou Akira (Osaka University, Japan) and were backcrossed with C57BL/6 wild-type (WT) mice for more than six generations.[21] Heterogenic mice were mated, and littermates used for experiments. All animal studies were conducted following the guidelines established by the Committee on the Ethics of Animal Experiments of the Kyungpook National University. Bone morphometric parameters and micro-architectural properties of the femur were determined using a micro-computed tomography (µCT) system (Quantum FX micro-CT; PerkinElmer, Waltham, MA, USA). Analysis of bone histology was performed as previously described.[22]
2. Osteoclast generation
Osteoclasts were derived from mouse bone marrow cells as previously described.[20] Briefly, whole bone marrow cells were cultured in alpha-minimal essential medium (MEM) containing 10% fetal bovine serum (FBS) with M-CSF for 3 days. The adherent cells (bone marrow macrophages [BMMs]) were used as osteoclast precursors. To differentiate cells into osteoclasts, BMMs were cultured with M-CSF (10 ng/mL) and RANKL (20 ng/mL) for 4 days.
3. TRAP staining
Cells were fixed in 4% paraformaldehyde and subsequently stained for TRAP activity with a 0.1 M acetate solution (pH 5.0) containing 6.76 mM sodium tartrate, 0.1 mg/mL naphthol AS-MX phosphate, and 0.5 mg/mL Fast Red Violet. Multinucleated cells positive to TRAP expression were identified as osteoclasts.
4. Proliferation and apoptosis assays
BMMs were cultured with various concentrations of M-CSF for 3 days. Subsequently, bromodeoxyuridine (BrdU) was added to the culture medium and incubated for 4 hr. Proliferation assay was performed using a cell-proliferation Biotrak ELISA system (GE Healthcare/Amersham, Freiburg, Germany). Apoptosis assay was conducted using the Cell Death Detection ELISA Kit (Roche Diagnostics, Mannheim, Germany), to detect cytoplasmic histone-associated DNA fragmentation.
5. Real-time quantitative polymerase chain reaction (PCR)
Real-time quantitative PCR was performed using the SYBR Green dye in the Applied Biosystem (ABI) 7500 real-time PCR machine (Applied Biosystems, Foster City, CA, USA). Primers were designed using Primer Express software (Applied Biosystems). The amplification reaction was performed for 40 cycles with denaturation at 95℃ for 10 min followed by annealing at 95℃ for 15 sec and extension and detection at 60℃ for 1 min. The following primers were used: c-Fos, 5'-AGGCCCAGTGGCTCAGAGA-3' and 5'-GCTCCCAGTCTGCTGCATAGA-3'; NFATc1, 5'-ACCACCTTTCCGCAACCA-3' and 5'-TTCCGTTTCCCGTTGCA-3'; TRAP, 5'-TCCCCAATGCCCCATTC-3' and 5'-CGGTTCTGGCGATCTCTTTG-3'; and LCN2, 5'-CACAGGTATCCTCAGGTACAGAGCTA-3' and 5'-GGAAAAATACCATGGCGAACTG-3'.
6. Western blotting and antibodies
Cells were harvested after washing with ice-cold phosphate buffered saline (PBS) and then lysed in radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris (pH 7.4), 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, 1 µg/mL pepstatin, and 1 µg/mL aprotinin. Whole cell lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was probed with specific antibodies, and immuno-reactivity was detected using enhanced chemiluminescence reagents (ECL Plus; Amersham Biosciences, Piscataway, NJ, USA). Antibodies against phospho-c-Fms, phospho-inhibitor of kappa B (IκBα), phospho-c-Jun, phospho-p38, phospho-ERK, phospho-Akt, p38, ERK, and Akt were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies for c-Fms and c-Fos were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody for NFATc1 was purchased from BD Pharmingen (San Diego, CA, USA).
RESULTS
1. LCN2 deficiency enhances osteoclast formation
Our previous study demonstrated that LCN2 regulates osteoclast formation.[20] In the present study, we used LCN2-deficient (LCN2-/-) mice, to further investigate the role of LCN2 in osteoclast development. BMMs derived from WT and LCN2-/- mice were cultured for 4 days in osteoclastogenic media containing M-CSF and two different concentrations of RANKL. As expected, RANKL treatment of WT BMMs increased the number of TRAP-positive multinucleated cells in a dose-dependent manner (Fig. 1A, B). Compared with WT counterparts, LCN2 deficiency resulted in a significantly increase of osteoclast numbers in both RANKL concentrations (Fig. 1A, B).
2. LCN2 deficiency increases proliferation of osteoclast precursors
The rate of precursor proliferation and apoptosis are important factors in determining osteoclast numbers. Therefore, increased osteoclast formation, due to LCN2 deficiency, could be caused by greater proliferation of osteoclast precursors and/or decreased apoptosis. To investigate whether LCN2 deficiency affects these parameters, we first assessed the apoptotic rate of WT and LCN2-/- BMMs by a DNA fragmentation assay with ELISA. We found that apoptotic rates are comparable in both WT and LCN2-/- cells (Fig. 2A), demonstrating that accelerated osteoclast formation is not caused by the decreased cell death seen in LCN2-/- BMMs. Next, we examined the impact of LCN2 deficiency on the proliferation of osteoclast precursors. For this purpose, we cultured WT and LCN2-/- BMMs for 3 days in the presence of the various concentrations of M-CSF. As previously reported, M-CSF increased cell growth in a dose-dependent manner (Fig. 2B). In LCN2-/- BMMs, BrdU incorporation was markedly increased at all concentrations of M-CSF (Fig. 2B). Thus, LCN2 deficiency affects proliferation of osteoclast precursors, but not their survival, thereby resulting in enhanced osteoclast formation.
3. LCN2 deficiency accelerates the expression and activation of c-Fms
M-CSF promotes the proliferation of osteoclast precursor cells by binding to c-Fms, its unique tyrosine kinase receptor. Since c-Fms plays a critical role in BMM proliferation, we investigated whether LCN2 deficiency influences c-Fms expression. As shown in Figure 2C, expression of c-Fms was markedly increased in LCN2-/- BMMs, when compared to WT cells. Accordingly, the phosphorylation of c-Fms in response to M-CSF was enhanced in LCN2-/- BMMs. Binding of M-CSF to c-Fms promotes the activation of downstream signaling pathways. Hence, we assessed M-CSF-stimulated MAPK and PI3K/Akt activation and established that ERK phosphorylation was significantly increased in LCN2-/- BMMs (Fig. 2C). On the other hand, phosphorylation of Akt was only modestly increased in LCN2-/- BMMs.
4. LCN2 deficiency enhances RANKL-induced c-Fos and NFATc1 expression
Based on our observations, LCN2 deficiency increases osteoclast formation. Therefore, we examined whether LCN2 deficiency influences the RANKL-induced expression of c-Fos, a pivotal transcription factor for osteoclast differentiation. LCN2 deficiency accelerated the RANKL-mediated induction of c-Fos mRNA, when compared to WT control (Fig. 3A). In concordance with the mRNA increase, LCN2 deficiency enhanced RANKL-induced c-Fos protein levels (Fig. 3B). Since NFATc1 acts as a downstream target of c-Fos, we then examined the impact of LCN2 deficiency on the RANKL-dependent induction of NFATc1, another transcription factor essential for osteoclastogenesis. As shown in Figure 3, LCN2 deficiency increased both mRNA and protein expression of NFATc1 in response to RANKL. Mirroring the increased expression levels of NFATc1, expression of osteoclastogenic markers such as TRAP and cathepsin K, was also enhanced as a result of LCN2 deficiency (Fig. 3A, B). We also observed that LCN2 receptors, 24p3R and megalin were expressed in both WT and LCN2-/- cells (Fig. 3C).
5. LCN2 deficiency increases RANKL-induced IκBα and c-Jun phosphorylation
RANKL controls osteoclastogenesis through the activation of downstream signaling pathways such as NF-κB and MAPKs. To determine the mechanism by which LCN2 regulates osteoclast differentiation, we examined the impact of LCN2 deficiency on RANKL signaling cascades. BMMs from WT and LCN2-/- mice were serum-starved and exposed to RANKL, to then perform an immunoblotting assay. While phosphorylation of p38 MAPK was not affected by LCN2 deficiency, we observed a marked enhancement of IκBα and c-Jun phosphorylation in LCN2-/- BMMs following RANKL stimulation (Fig. 3D).
6. In vivo analysis of LCN2-/- mice
To investigate the role of LCN2 in vivo, we characterized the bone phenotype of LCN2-/- mice. Interestingly, micro-computed tomography analysis of the distal femur from 2-month-old LCN2-/- and WT mice showed no significant differences between strains (Fig. 4A). Both WT and LCN2-/- mice had similar bone volume per tissue volume (BV/TV), bone mineral density (BMD), trabecular number (Tb. N), and trabecular space (Tb. Sp) (Fig. 4B). Histological examination also revealed no apparent differences between WT and LCN2-/- mice (Fig. 4C). Similarly, serum levels of C-terminal telopeptides of type I collagen (CTX-1) were unaltered; suggesting that in vivo basal osteoclast development in LCN2-/- mice is normal (Fig. 4B). Together, these results suggest that LCN2 deficiency does not significantly impact osteoclast formation in vivo under normal physiological conditions.
DISCUSSION
We previously demonstrated that LCN2 is expressed in osteoclast lineage cells and that its expression is regulated during osteoclastogenesis. Subsequent studies, including ectopic expression and recombinant LCN2 protein treatment, revealed that LCN2 suppresses the proliferation and differentiation of osteoclast precursors. In the present study, we used a genetically engineered LCN2-/- mice and their WT littermates, to confirm that LCN2 negatively regulates in vitro osteoclast formation in response to M-CSF and RANKL.
It has been shown that LCN2 plays a critical role in cell proliferation. LCN2 inhibits the growth of bacteria through the formation of a complex with bacterial siderophore.[10, 11] Furthermore, a previous study using LCN2-/- mice, revealed that LCN2 suppresses bacterial growth, thereby protecting host against bacterial infection.[12] In the context of hematopoiesis, LCN2 also has been reported to suppress the growth of erythroid and monocyte/macrophage progenitors, in both mice and humans.[1617] Consistent with these findings and results from our previous work,[20] LCN2 deficiency accelerates the proliferation of osteoclast precursor cells, BMMs. On the other hand, LCN2 deficiency did not influence the apoptosis of osteoclast progenitors.
Osteoclast number is determined by proliferation, differentiation, and apoptosis rates of osteoclast lineage cells. The terminally differentiated osteoclasts are non-proliferative, but their precursors can proliferate under the control of M-CSF and its receptor c-Fms. The importance of M-CSF in osteoclast biology has been well demonstrated in previous studies. The naturally occurring op/op mice, which cannot express functional M-CSF due to a mutation in the csf1 gene, develop osteopetrosis due to complete absence of macrophages and osteoclasts.[2324] The critical role of c-Fms in osteoclast development was demonstrated using genetically modified mice. Mice-deficient csf1r, the gene coding for c-Fms, exhibit the same major phenotype as the op/op mice as a result of a significant decrease in macrophages and lack of mature osteoclasts.[25] In the present study, we observed that LCN2 deficiency enhances the expression and activation of c-Fms. Thus, accelerated proliferation of LCN2-/- BMMs is consistent with the increased expression of c-Fms protein. Consequently, downstream signaling pathways such as ERK and Akt, were increased due to LCN2 deficiency. Collectively, studies show that LCN2 suppresses M-CSF-induced precursor proliferation through the regulation of c-Fms expression, which in turn results in defective osteoclast formation.
c-Fos and NFATc1 are key modulators of RANKL-induced osteoclastogenesis.[92627] Our previous work demonstrates that overexpression or treatment with recombinant LCN2 attenuates RANKL-induced c-Fos and NFATc1 expression. Consistent with our previous results, we observed that c-Fos and NFATc1 induction was significantly enhanced in BMMs and osteoclasts derived from LCN2-/- mice. Consequently, NFATc1 leads to the increased expression of TRAP and cathepsin K, which are required for osteoclast differentiation and function.
Stimulation of RANKL activates MAPK signaling pathways including c-Jun N-terminal kinase (JNK), ERK, and p38. These MAPKs signaling pathways then activate AP-1 components including c-Fos and c-Jun, which in turn enhance osteoclast differentiation. NF-κB activation by RANKL is another pivotal event for osteoclastogenesis. Consistent with accelerated osteoclast differentiation, phosphorylation of IκBα and c-Jun was increased in LCN2-/- BMMs. On the other hand, activation of p38 was not affected by LCN2 deficiency. Notably, our previous work revealed that addition of LCN2 inhibits IκBα phosphorylation, nuclear translocation of the NF-κB subunit p65, and NF-κB transcriptional activity in response to RANKL. Thus, suppression of RANKL-mediated NF-κB activation is likely to be an important component of anti-osteolastogenic properties of LCN2.
Our study provides clear evidence that LCN2 deficiency increases osteoclast formation in vitro. However, in contrast to in vitro data, in vivo basal bone phenotype is normal in LCN2-/- mice. One possible explanation is that osteoclast formation, in vitro, reflects an activated state. Likewise, mice lacking Lyn,[28] Fyn,[29] and NF-κB-inducing kinase (NIK),[30] also show no basal phenotype in vivo, despite the profound impact on osteoclast number in vitro. As these mice represent in vivo phenotype in an activated state, but not in a basal state, we speculate that LCN2 may also play a role in the accelerated osteoclast development that occurs under a variety of pathological conditions, including osteoporosis and rheumatoid arthritis. However, whether this is the case in the context of LCN2 needs to be further investigated. Nonetheless, a recent study demonstrated that LCN2-/- mice develop severe serum-induced arthritis when compared to WT mice.[31] The study demonstrated, through histological examination, that mice paws of LCN2-/- mice have significant bone erosion compared to their WT counterparts.[31] These studies indicate that LCN2 has a protective role in the development of inflammatory diseases and in the preservation of bone homeostasis. In this regard, our study provides another piece of evidence that LCN2 functions as a negative regulator of osteoclast development, when induced by M-CSF and RANKL.
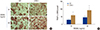



 XML Download
XML Download