

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bone homeostasis is regulated by the process of bone remodeling, which maintains a delicate balance between bone resorption by osteoclasts and bone formation by osteoblasts. Disturbance of the balance of differentiation or activation between osteoclasts and osteoblasts leads to various skeletal disorders, such as postmenopausal osteoporosis, rheumatoid arthritis, and some osteolytic metastatic cancers.[1] Therefore, restoration of unbalanced bone remodeling is a critical goal of pharmacological interventions for excessive bone destruction. Chemical inhibitors of osteoclastic bone resorption have primarily been used for the treatment of excessive bone destruction. However, bone-forming drugs would be helpful for patients who have already lost considerable amounts of bone.[2]
Histone deacetylases (HDACs) are well known to mediate the epigenetic regulation of gene expression by removing acetyl groups from lysine residues on histone core proteins, leading to chromatin condensation and transcriptional repression.[3] The acetylation, a key posttranslational modification of many proteins, plays an important role in the modulation of numerous critical intracellular pathways, including chromatin remodeling and transcription, microtubule dynamics, aging, and differentiation.[4] Because HDACs are recognized as a potential target for cancer treatment, many HDAC inhibitors have been entered into clinical trials.[5] To date, 18 HDACs have been identified in mammalian cells and are divided into subclasses: class I HDACs, class IIa HDACs, class IIb HDACs, class III HDACs, and class IV HDAC. In addition to histones, HDACs also affect the acetylation status of nonhistone proteins such as p53, signal transducer and activator of transcription 1 (STAT1), and tubulin.[6] Several HDAC inhibitors including SCOP402, CHAP27, and trichostatin A increase runt-related transcription factor 2 (Runx2) acetylation, potentiate bone morphogenetic protein-2 (BMP-2)-stimulated osteoblast differentiation, and increase bone formation.[7] Furthermore, the critical roles of several HDACs, including HDAC1, HDAC3, HDAC4, HDAC5, HDAC6, HDAC8, and Sirtuin1, have been identified in the regulation of osteoblast differentiation.[8] Some HDAC inhibitors including valproic acid and sodium butylate have been shown to induce osteogenic differentiation of human mesenchymal stem cells and several osteogenic cell lines.[9,10]
We previously showed that MS-275-mediated osteogenic potential induced a human DNA helicase called Dhs36, which was definitely required for induction of tissue-nonspecific alkaline phosphatase (TNAP).[11] The physical interaction between Dhs36 and a subset of HDACs seemed to be of importance. However, the presumable signal transduction resulting in osteoblastogenesis elicited by MS-275 seemed to be independent of that of BMP-2.
Suberoylanilide hydroxamic acid (SAHA; vorinostat) is the first HDAC inhibitor approved by the Food and Drug Administration to treat tumors and is undergoing rapid clinical trials in various solid tumors.[12] Here, we report that SAHA significantly inhibited the onset of osteoporosis at much lower concentrations than in a previous report.[13] More interestingly, SAHA per se was incapable of promoting osteoblastogenesis in vitro, unlike MS-275, but was significantly synergistic with BMP-2 at low concentrations for inducing alkaline phosphatase (ALP), which might be mediated by stabilization of Runx2 by retaining acetylation. Consistent with this in vitro study, administration of SAHA into mice inhibited a significant loss of trabecular bone in a soluble receptor activator of nuclear factor-kappa B ligand (sRANKL)-induced osteoporotic mouse model.
METHODS
1. Cell cultures and reagents
C2C12 cells and HEK-293T cells were maintained in Dulbecco's modified Eagle's medium with 10% heat-inactivated fetal bovine serum supplemented with 50 units/mL penicillin and 50 µg/mL streptomycin. Penicillin, streptomycin, alpha-minimal essential medium (α-MEM), and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA, USA). Recombinant human BMP-2, soluble human RANKL, and human fibroblast growth factor fibroblast growth factor-2 (FGF-2) were obtained from PeproTech (London, UK). Human parathyroid hormone (PTH [1-34]) and SAHA were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2. Animal experiments
All animal procedures were approved by the animal care committee of the Institute of Laboratory Animal Resources of Seoul National University. To induce rapid bone loss, sRANKL (1 mg/kg of body weight) or phosphate buffered saline (PBS) was injected intraperitoneally at 24-hr intervals for 2 days into 7-week-old female ICR mice (n=7 mice/group) as described previously.[11] Two days after the last sRANKL injection, human PTH (1-34) (160 µg/kg), SAHA (0.25 mg, 1 mg, 10 mg/kg), or vehicle (50% dimethylsulfoxide [DMSO]) was injected subcutaneously for 10 consecutive days. The mice were sacrificed on day 14, and the right femora were dissected, cleaned of soft tissue, fixed in 4% paraformaldehyde, and then analyzed by micro-computed tomography (CT) scanning. Micro-CT was performed with an SMX-90CT system (Shimadzu, Kyoto, Japan; 90 kVp, 109 mA, and 180-ms integration time). Scans were then integrated into 3-dimensional voxel images (1,024-pixel×1,024-pixel matrices). All bone images were reconstructed with the VG studio MAX 1.2.1 program (Volume Graphics, Heidelberg, Germany) by use of standard procedures. The regenerated bone volume/tissue volume (BV/TV; %), trabecular thickness (Tb.Th; µm), and trabecular separation (Tb.Sp; µm) were calculated and expressed with TRI/3D-VIE (RATOC System Engineering, Tokyo, Japan) according to standard formulas and nomenclatures.
3. Osteogenic differentiation and ALP staining
C2C12 cells and 293T cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (FBS), 50 units/mL penicillin, and 50 µg/mL streptomycin. To induce osteogenic differentiation, C2C12 cells (5×104 cells/well) were plated in 48-well plates and cultured until 90% confluent. SAHA and BMP-2 were then added at the indicated concentrations. Cells were stained with an ALP kit (Sigma-Aldrich, Cat. no. 86R-1KT) according to the manufacturer's instructions.
4. Luciferase assay
For the luciferase assay, C2C12 cells were transfected in 12-well plates with 300 ng/well of the p6xOSE2-luc construct by using Hyperfect transfection reagent (Qiagen, Valencia, CA, USA) as described previously.[14] The cells were treated with SAHA at the indicated concentrations with FGF-2 as a control at 24 hr after transfection. Luciferase activity was measured by using the dual-luciferase assay system (Promega, Madison, WI, USA) in a Dynex luminometer.
5. Immunoprecipitation and immunoblotting
293T cells were washed in cold PBS and lysed on ice in lysis buffer (20 mM Tris, pH 7.5, 50 mM NaCl, 0.1% NP-40, 2 mM EDTA, and protease inhibitors), incubated on ice for 30 min, and then cleared by centrifugation at 13,000×g for 15 min. A total of 500 µg of protein was incubated with 5 µg of anti-acetylated lysine antibody overnight with rotation. An amount of 40 µL of protein A-agarose beads (Sigma Chemical Co., St. Louis, MO, USA) was then added and incubated with rotation for 2 hr at 4℃. Immunoprecipitates were washed extensively 3 times in lysis buffer and bound proteins were resolved by SDS-PAGE. The immunoblotting procedure with anti-myc antibody (Sigma-Aldrich, St. Louis, MO, USA) was performed as described previously.[15]
RESULTS
1. SAHA drives the transcription of the Runx2 promoter in vitro
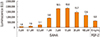
Runx2 plays a central role in osteoblastogenesis.[16] To see if the Runx2 promoter responded to SAHA, a transient reporter assay was conducted by using 6XOSE as previously described.[14] As shown in Figure 1, luciferase activity was generated in a dose-dependent manner, with maximal activity seen with 10 µM SAHA. The EC50 was calculated as 1.0 µM. At concentrations over 10 mM, luciferase activity declined but was higher than that with treatment with FGF-2 (10 ng/mL).These findings suggested that a biological entity associated with SAHA would be a bonafide activator of osteoblastogenesis.
2. The osteogenic activity of SAHA is synergized by BMP-2
To determine whether SAHA inhibited osteoblastogenesis, we treated C2C12 cells, a mouse premyoblastic cell line, with SAHA in various concentrations from 0.1 µM to 30 µM and measured ALP activity. Interestingly, as shown in Figure 2, SAHA per se was unable to induce osteoblastogenesis regardless of the concentration used. Because SAHA can trans-activate the Runx2 promoter, this inability was unexpected. We hypothesized that SAHA might need a catalyst for osteoblastogenesis and therefore added BMP-2 at a low concentration of 30 ng/mL. Indeed, the addition of BMP-2 synergized the osteoblastogenic ability of SAHA treatment at 1.0 µM or 3 µM, with resultant activity higher than that with BMP-2 alone (Fig. 2).
3. SAHA stabilizes an acetylated form of Runx2
The loss of the either allele for Runx2 leads to cleidocranial dysplasia, which is characterized by severe bone loss and deformation.[17] The acetylation of Runx2 by treatment with BMP-2 is responsible for protein stability. Furthermore, HDAC inhibitors can attenuate the deacetylation by HDAC, giving rise to perpetuation of differentiation of osteoblasts. We therefore established an in vitro stability test with regard to acetylation of Runx2. 293T cells were transfected with 3XMyc-tagged Runx2 and treated with SAHA in various concentrations. The cell lysates were immunoprecipitated with an acetylation-specific antibody or a c-Myc antibody. As shown in Figure 3, Runx2 was more acetylated with SAHA treatment at 1.0 µM or 10 µM. The degree of over-expression of Runx2 was confirmed by anti-Myc. The blot was normalized by anti-β actin. Consistent with our hypothesis, SAHA stabilized the acetylation of Runx2.
4. SAHA can attenuate sRANKL-induced osteoporosis in vivo
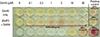
ICR mice were administered SAHA at different concentrations or vehicle in conjunction with sRANKL. After 4 days, PTH or SAHA were administered and the mice were sacrificed. Bone-promoting activities were measured with µCT. As shown in Figure 4, compared with sRANKL alone, which clearly promoted bone resorption, PTH or SAHA significantly blocked osteoporosis. The dose of 1 mg/kg gave rise to the most effective activity. Using trabecular bones, we measured several bone parameters such as bone volume, thickness, and bone number. Consistent with the µCT data, these parameters were significantly increased with administration of SAHA, and the dose of 1 mg/kg exhibited the most effectiveness. Taken together, these results suggest that SAHA is an efficient blocker of osteoporosis in vivo and a stabilizer of acetylated Runx2 in vitro; however, the effect of SAHA seems to depend on BMP-2.
DISCUSSION
We previously showed that MS-275 exhibited potent osteogenic potential via induction of a DNA helicase called Dhs36, which then trans-activates the proximal promoter of TNAP.[11] The induction seemed to require some subsets of HDACs with Dhs36. However, BMP-2 signaling did not seem to be involved in the transcriptional activation of TNAP by Dhx36 and the aiding of the physical interaction of Runx2 with Dhx36 by HDACs. The induction of Dhx36 did not seem to be affected by BMP-2 and MS-275 was unable to induce Runx2, which suggested that the signal transduction pathways of the MS-175-mediated osteogenic potential may be distinct from those of BMP-2. The presence or absence of HDAC enzymatic activity was not required for the induction of ALP, but retaining HDAC protein was essentially required. In contrast, the osteogenic potential of SAHA depended on BMP-2 in vitro, because SAHA per se was unable to induce ALP in C2C12 cells and was only synergized in the presence of low concentrations of BMP-2. Because Runx2 is believed to play a central role in osteoblastogenesis,[18] we tested whether SAHA affected the stability of Runx2 via acetylation. We observed that SAHA led to a significant degree of acetylation of Runx2. This suggests that the signaling pathways of SAHA in C2C12 cells, which led to an acquisition of osteogenic potential, are different from those of MS-275. Thus, a rational question was whether SAHA could promote anti-osteoporotic activity in vivo. Indeed, we showed that SAHA substantially inhibited the osteoporosis induced by the administration of sRANKL. Although we have not tested whether this is the case in BMP-deficient mice, our in vitro data suggest that the osteogenic effect of SAHA might be mediated through endogenous BMP-2. This possibility remains to be further elucidated. Given that the maximal effective concentration of SAHA in vivo was 1 mg/kg, this may be a safe concentration to use in future preclinical studies.
The results of the present study also suggest that HDACs exploit many diverse signaling pathways leading to osteogenesis. In agreement with this conjecture, a study administering SAHA into C57BL/6J mice showed a loss of trabecular bone in the distal femur of mice, mainly through a decrease in immature osteoblast number.[13] We postulate that this discrepant in vivo results are related to the frequency and dose of SAHA treatment. Because the mice were treated with SAHA of a high dose at 100 mg/kg and longer duration for 3 or 4 weeks, such an outcome might manifest as a considerable toxicity in the animals. In another study, epigenomic alteration by SAHA was responsible for the promotion of osteoblastogenesis by use of the committed osteoblast cell line MC3T3,[19] which suggests that the intrinsic degree of osteoblastogenesis might also be a determining factor in the response to SAHA and other HDACs. A great deal of uncertainty remains concerning the osteogenic potential of SAHA. We observed that SAHA could restore acute osteoclastogenesis in an sRANKL-induced mouse bone loss model. This study may reflect that a diverse array of HDAC signaling pathways exist and are fine-tuned in response to the differentiation fate of osteoblast stem cell progenitors and the cytokine milieu in the bone marrow niche. In summary, we found a novel osteogenic potential associated with SAHA, which, unlike that of MS-275, may be dependent on BMP-2.


 XML Download
XML Download