

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
In the past 4 years, genome-wide association studies (GWASs), assaying hundreds of thousands of single nucleotide polymorphisms (SNPs) in thousands of individuals, have identified a number of promising genetic variants that are associated with osteoporosis and related traits. The first published whole genome sequencing study for osteoporosis and fractures identified a novel rare nonsense mutation. This article reviews the current status of GWASs of osteoporosis and their meta-analyses with an emphasis on prominent results, approaches, and problems with these studies. The major findings of the first whole genome sequencing study for osteoporosis and fractures are also briefly discussed. We focus primarily on bone mineral density (BMD), the most important risk trait for osteoporosis, and on osteoporotic fracture (OF), the most severe clinical outcome of osteoporosis. We address how to interpret the discordance of research findings between individual GWASs and meta-analyses, and between different meta-analyses. The values and limitations of GWAS and meta-analysis are evaluated based on empiric and theoretical analyses. Finally, future directions using multi- and inter-disciplinary study strategies for genetic research of osteoporosis are discussed, with potentially significant implications for the general human genetics field.
GWASs AND META-ANALYSIS ON OSTEOPOROSIS AND OF
Osteoporosis is the most common metabolic skeletal disorder in humans. It predisposes people to fragility fractures and confers substantial morbidity and mortality, affecting over 200 million people worldwide.[1,2] Osteoporosis is mainly characterized by low BMD, a highly heritable trait with heritability ranging from 0.5 to 0.8.[3,4,5,6] OF, as an end-point clinical outcome of osteoporosis, also has moderate heritability, of approximately 0.5-0.7.[7,8] To date, GWASs and their meta-analyses have identified over 60 genes/loci associated with variations in BMD and more than 20 genes/loci associated with risk of OF. In addition, a most recently published whole-genome sequencing study identified a rare nonsense mutation novel within a novel gene LGR4 that was strongly associated with low BMD and OF.[9]
The majority of published GWASs have focused on BMD using SNP data. A recent review by Richards et al.[10] summarized the major findings from SNP-based GWASs, which will not be repeated here. Instead, we highlight prominent genes or loci identified in SNP based GWASs with a focus on consistency and inconsistency of results. We address issues related to interpretation of meta-analysis results and replication of study findings among GWASs and meta-analyses. A few GWASs have focused on genome wide analyses of copy number variants (CNV) and biological pathways. The major findings from these latter studies are briefly introduced and discussed.
1. GWASs based on SNPs
To date, a total of 19 GWASs have been published for osteoporosis. Of these, 14 are individual GWASs, and 5 are GWAS meta-analyses. The significant genes and loci identified from these studies, along with information regarding study design (e.g., individual GWASs or meta-analysis) and phenotypes (e.g., hip and/or spine BMD or OF) are summarized in tables 1 and 2.
1) GWAS meta-analyses
The Genetic Factors of Osteoporosis (GEFOS) consortium published two large-scale GWAS meta-analyses.[11,12] Their first meta-analysis (GEFOS-1), which included 19,195 subjects of European descent, identified 20 BMD loci.[11] Their second GWAS meta-analysis (GEFOS-2), the largest one to date in the bone field, included 32,961 individuals in the discovery phase and was replicated in 50,933 independent subjects.[12] The study subjects included Europeans and East Asians. GEFOS-2 identified a total of 56 BMD loci at the genome-wide significance level. Of these 56 loci, 32 were novel and the remaining 24 were genes/loci that were previously known to affect bone mass regulation and metabolism (e.g., receptor activator of nuclear factor (NF)-kappaB (κB) [RANK], RANK ligand [RANKL], and lipoprotein receptor-related protein 5 [LRP5]). Notably, multiple loci achieved highly significant associations in GEFOS-2, with the magnitude of P values reaching10-60 for 2 loci, 10-50 for 1 locus, 10-40 for 2 loci, 10-30 for 6 loci, 10-20 for 11 loci, and 10-10 for 27 loci. GEFOS-2 also revealed 14 loci associated with risk of fractures (Table 2). However, fractures used in the analyses were quite heterogeneous, and included hip, spine, and wrist, as well as other types of fractures. Due to the well-known genetic and non-genetic etiological heterogeneity underlying different types of fractures,[2,8,13,14,15,16] these study findings should be interpreted with caution, prior to independent validation in other samples with homogeneous fracture types.
Koller et al.[17] carried out a meta-analysis of GWASs restricted to premenopausal white women from four cohorts (n=4,061, aged 20 to 45 years), a subset of GEFOS-2, and identified two loci (wingless-type MMTV integration site family, member 16 [WNT16] and estrogen receptor 1 [ESR1]/C6orf97) influencing peak bone mass at the lumbar spine and femoral neck. Only 4 of the 56 loci detected in the joint female GEFOS analysis[12] were observed to have P values below 5×10-5 in this study.
Zhang et al.[18] conducted a three-stage GWAS meta-analysis in 27,061 study subjects. Stage 1 meta-analyzed seven GWA samples and 11,140 subjects for BMDs at the lumbar spine, hip and femoral neck, followed by a Stage 2 in silico replication of 33 SNPs in 9,258 subjects, and by a Stage 3 de novo validation of three SNPs in 6,663 subjects. Combining evidence from all the stages, two novel loci were identified at the genome-wide significance level: 14q24.2 (rs227425, P=3.98×10-13, SPARC related modular calcium binding 1 [SMOC1]) and 21q22.13 (rs170183, P=4.15×10-9, claudin 14 [CLDN14]). These two SNPs were also significant in GEFOS-2.[12] This study independently confirmed 13 previously reported loci.[18] Further gene expression analysis in osteogenic cells implied potential functional association of the two novel candidate genes (SMOC1 and CLDN14) in bone metabolism.
Most studies have focused on areal BMD (aBMD) obtained from a 2-dimensional projectional scan with dual energy X-ray absorptiometry (DXA). Although aBMD is the gold standard for diagnosing osteoporosis, it fails to provide a detailed information necessary to discern traits such as trabecular volumetric BMD (vBMD), cortical vBMD and bone microstructural parameters. Quantitative computed tomography (QCT) analysis has the advantage to reveal unique information about these bone traits. Paternoster et al.[19] published the first GWAS to identify genetic loci associated with cortical and trabecular bone microstructural parameters in European Caucasians. Their cortical vBMD GWAS meta-analysis (n=5,878) followed by replication (n=1,052) identified genetic variants in four separate loci (RANKL, rs1021188, P=3.6×10-14; LOC285735, rs271170, P=2.7×10-12; osteoprotegerin [OPG], rs7839059, P=1.2 × 10-10; and ESR1/C6orf97, rs6909279, P=1.1×10-9). The trabecular vBMD GWA meta-analysis (n=2,500) followed by replication (n=1,022) identified one locus reaching genome-wide significance (formin 2 [FMN2]/gremlin 2, DAN family BMP antagonist [GREM2], rs9287237, P=1.9×10-9). In addition, rs1021188 was associated with cortical porosity while rs9287237 was associated with trabecular bone fraction. The genetic variant in the FMN2/GREM2 locus was also associated with fracture risk in the MrOS Sweden cohort and GREM2 expression in human osteoblasts. Two of these (FMN2/GREM2 and LOC285735) are novel bone related loci, while the other three have previously been reported to be associated with aBMD. This study provided evidence that the genetic determinants of cortical and trabecular vBMDs differ. However, QCT has its limitations, including being not applicable to World Health Organization (WHO) definition of osteoporosis that is based on DXA measurement, being more expensive with a higher dosage of exposure to radiation and may not predict fractures better than DXA measurement.[20,21] Nevertheless, its advantages over DXA make QCT a complementary (but not necessarily replacement) approach to bone health assessment.[22]
Conducting these large-scale meta-analyses requires an extensive, concerted effort in collaboration and coordination among various research centers and groups, as well as centralization and standardization in data analyses. Thus, publication of these studies represents one of the most impressive achievements in the osteoporosis genetics field. Based primarily on the large samples involved in meta-analyses such as these, some researchers have begun to consider large meta-analyses as a gold standard for evaluating other individual GWASs, with an explicit assumption that validity of the findings from other studies of smaller samples needs to be confirmed in large meta-analyses such as GEFOS-2.[12]
Comparing the results of the two published GEFOS meta-analyses, it is interesting to note that several loci identified in GEFOS-1 (e.g., corticotropin releasing hormone receptor 1 [CRHR1] and histone deacetylase 5 [HDAC5]) were not significant in GEFOS-2, despite the fact that the number of samples of GEFOS-2 almost tripled the number in GEFOS-1, and the majority of samples used in GEFOS-1 were included in GEFOS-2.
Further comparing the results of these meta-analyses with individual GWASs, it is intriguing that some significant loci identified in meta-analyses were found to be significant at the genome-wide level in individual GWASs, while others were not. Likewise, some significant loci identified in individual GWASs were replicated in meta-analyses, while many others were not. We will highlight some of these consistencies and inconsistencies in the following paragraphs and discuss how to interpret these findings.
2) Individual GWASs
(1) Single ethnicity
The first two individual GWASs were performed in human subjects of European ancestry [23,24] using the data from TwinsUK/Rotterdam and deCODE Genetic studies. These two studies identified a total of five loci at the genome-wide significance level (P<5×10-8). The associated genes, including LRP5, OPG, RANK, RANKL and ESR1, are well known for their importance to bone and mineral metabolism, and have been well established in previous candidate gene studies.
The efficiency of extreme-truncate selection design for quantitative trait association studies has been well established.[25] Duncan et al. adopted this design by focusing on postmenopausal Caucasian women with either extremely high or low hip BMD in the discovery stage.[26] Although the sample size of the discovery phase was moderate (1,955 subjects in discovery phase), the power of the study was still high due to the extremely discordant sample design. In addition to replicating 21 of 26 known BMD-associated genes, they identified six new associations in/around the genes chloride channel, voltage-sensitive 7 (CLCN7), UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3), integrin-binding sialoprotein (IBSP), latent transforming growth factor beta binding protein 3 (LTBP3), R-spondin 3 (RSPO3), and SRY (sex determining region Y)-box 4 (SOX4). Some of these genes (e.g., LTBP3) were not significant at the genome-wide significance level in GEFOS-2, although the samples used in this study were included in GEFOS-2.
Organic integration of information from different levels and disciplines may help identify genetic variants of modest or moderate effects. By integrating GWAS in Caucasians and expression signature profiling of various human tissues, Hsu et al.[27] identified three novel genes, member of RAS oncogene family (RAP1A), TBC1 domain family, member 8 (with GRAM domain) (TBC1D8), and oxysterol binding protein-like 1A (OSBPL1A), for BMD, and replicated a well-known gene, OPG. They also prioritized 16 suggestive genome-wide significant candidate genes, among which two candidate genes, G protein-coupled receptor 177 (GPR177) and SOX6, were replicated in GEFOS-2. The samples used in this study (mainly from the Framingham Osteoporosis Study) were included in GEFOS-1 and GEFOS-2. Therefore, it does not represent an independent replication. The three identified novel genes (i.e., RAP1A, TBC1D8, and OSBPL1A) were not significant at the genome-wide level in either GEFOS-1[11] or GEFOS-2.[12] Gene expression profiling analyses in this study were conducted in lymphocytes and the liver whose expression profiles may not be directly related to bone. Moreover, it is the differential gene expression with regard to osteoporosis related phenotypes, not simply expression of genes, which is more clinically relevant to risk of osteoporosis and OF.[28,29]
(2) Multi-ethnicities
To investigate potential population generality/specificity of genes/loci, several GWASs were conducted in populations of multiple ethnicities. Xiong et al.[30] used a US Caucasian cohort as the discovery sample followed by replication analyses in independent populations including those of African (from the Tobago study) and Asian ancestry. Two novel genes, ADAM metallopeptidase with thrombospondin type 1 motif, 18 (ADAMTS18) and transforming growth factor, beta receptor III (TGFBR3), were identified at the genome-wide significance level. These two genes were found to be significant in other individual GWASs conducted in completely independent samples by independent investigators in premenopausal US Caucasian women[31] and in postmenopausal women with extremely discordant BMD values (28). However, neither of these genes was found to be significant in the two GEFOS meta-analyses.[11,12]
Focusing on subjects of a specific gender and/or age range may reduce the potential confounding effects of heterogeneity, and empirically increase the power of a study. Koller et al.[31] conducted a GWAS in 1,524 premenopausal US Caucasian women aged 20-45 years and 669 African American premenopausal women aged 20-44 years. A novel gene, catsper channel auxiliary subunit beta (CATSPERB), was identified as being significant for peak BMD. Despite the fact that samples from this study were included in GEFOS-2,[12] CATSPERB was not found to be significant in the meta-analyses.
(3) Children
Although the majority of GWASs have been focused exclusively on adults, Timpson et al.[32] published the first GWAS in 1,518 children from the Avon Longitudinal Study of Parents and Children. They identified association with BMD at the Osterix (SP7) gene locus, a transcription factor responsible for regulating osteoblast differentiation. In another study, Medina-Gomez et al.[33] identified a novel gene, Wnt16 that was associated with total body and skull BMD variation in children and adults, which suggested a role in peak bone mass accrual which may impact the risk of osteoporosis later in life. Interestingly, this locus was also associated with cortical bone thickness, wrist BMD, bone strength, and risk of forearm fracture in adults,[34] peak bone mass in premenopausal women,[17] and BMD and fracture in elderly individuals of European descent.[12]
(4) OF
The majority of the published GWASs of osteoporosis have been focused on BMD, a major risk factor for OF. However, OF is an important clinical end-point of osteoporosis, and is the most clinically relevant trait. Earlier studies have demonstrated a high genetic determination for OF risk,[13] and have also shown that BMD and fracture may have different genetic determination,[7,8] indicating the necessity to perform genetic studies on OF per se. Over the past several years, OF, as an independent study phenotype, has received increased attention in the bone genetics field.[35,36] Currently, a major problem facing genetic studies of OF is the difficulty in recruiting sufficiently large and homogeneous samples of the same OF type.
The first published GWAS for OF used 700 elderly Chinese Han subjects (350 with hip OF and 350 healthy matched controls) in the discovery phase, followed by replication in an independent Chinese sample containing 390 cases with hip OF and 516 controls.[37] A novel gene, aldehyde dehydrogenase 7 family, member A1 (ALDH7A1), was identified to be significant for hip OF. Further analyses confirmed its relevance to hip BMD in both Chinese (sample size, 2,955) and Caucasian (sample size, 7,007) populations. Although the discovery sample size was relatively small, the study was conducted in a homogeneous sample of the same ethnicity and the same type of carefully clinically adjudicated OF, which may empirically increase the power of gene identification. Notably, ALDH7A1 was not found to be significant in GEFOS-2.
Hwang et al.[38] published a GWAS for OF in East Asian populations. They first conducted a GWAS in the discovery cohort which included 288 cases and 1,139 controls, followed by a two-stage de novo replication analyses which included 462 cases and 1,745 controls in stage 1 and 369 cases and 560 controls in stage 2. A new locus associated with OF (rs784288 in the MECOM gene) showed genome-wide significance (P=3.59×10-8; OR 1.39). Further RNA interference analysis revealed that a MDS1 and EVI1 complex locus (MECOM) knockdown suppresses osteoclastogenesis.
An alternative approach for studying the genetic determination of OF is to first identify BMD loci, and then test the relevance of those loci to OF. This approach has been used for individual GWASs[23,24,30] and for GEFOS-2 (Table 2).[12] Using this approach, GEFOS-2 identified 14 BMD loci that were associated with risk of OF. One potential problem with the GEFOS-2 OF analyses is that it combined OFs from a variety of clinically heterogeneous types of OFs, thus departing from the assumption of homogeneity made by meta-analysis for the phenotype. From perspective of bone biology, the proportions of cortical and cancellous bone differ at the different sites in the skeleton (e.g., hip, spine and wrist) where OFs frequently occur.[39] The differences in behavior of bone at the different sites are most likely caused by the different environments of the bone cells in cortical or cancellous bone.[39] Moreover, each type of fracture has its own unique risk factors.[2] For instance, fall is a major risk factor for hip and wrist fractures; however, it contributes little to the risk of vertebral fracture. Epidemiology data also show that the prevalence of fracture at different sites is different in terms of age, gender and ethnicity.[2,40] So far, the majority of the genetic variants identified by GWASs or candidate gene studies for BMD at different skeletal sites do not overlap.[5,10] Given these evidences, it is likely that different skeletal sites may not share much common genetic component. Therefore, the mixing of phenotypes in GEFOS-2 meta-analysis complicates the interpretation of results, as increasing sizes of non-homogeneous samples may lead to false positive/negative results. Although meta-analysis can be very powerful, its application must be executed with caution.
2. GWASs based on CNVs
SNP-based GWASs, including GEFOS meta-analyses, have discovered genes/loci which, collectively, account for less than 6% of the risk for developing osteoporosis. Therefore, it is important to explore the possibility that some of the remaining undiscovered genetic factors that influence risk of osteoporosis are due to genomic mechanisms other than individual nucleotide changes, such as CNVs.
CNV is a common type of genomic variability, with variations in the size of DNA fragments ranging from 1 kilobase to several megabases. CNVs, which may include duplications or deletions, can influence gene expression by disrupting coding sequences, perturbing long-range gene regulation, or altering gene dosage, and these effects could contribute to phenotypic variations or disease risk.[41]
A CNV-based GWAS for osteoporosis was conducted by Yang et al.[42] in 700 elderly Chinese individuals comprising 350 cases with homogeneous hip OF and 350 matched controls. In that study, CNV 4q13.2 was strongly associated with OF. Further validation experiments identified a deletion variant of UDP glucuronosyltransferase 2 family, polypeptide B17 (UGT2B17) in CNV 4q13.2, and the association between CNV of UGT2B17 and OF was replicated in an independent Chinese sample containing 399 cases with hip OF and 400 controls.[42] Despite the relatively small sample sizes, the CNV focused GWAS identified novel genes/loci that were otherwise missed in SNP-based GWASs.
Oei et al.[43] performed a genome-wide CNV analysis in 5,178 individuals from Netherlands, including 809 OF cases, and followed by in silico lookups and de novo replication in several independent samples. A rare (population prevalence 0.14%) 210 kb deletion located on chromosome 6p25.1 was associated with the risk of fracture. The prevalence of this deletion showed geographic diversity, being absent in samples from Australia, Canada, Poland, Iceland, Denmark, and Sweden, but present in the Netherlands (0.34%), Spain (0.33%), USA (0.23%), England (0.15%), Scotland (0.10%), and Ireland (0.06%). Larger and geographically restricted studies are needed to confirm this regional association.
3. GWASs based on biological pathways
Conventional GWASs consider the effects of individual genetic markers independently, and focus only on genes or markers of top-ranking statistical significance. However, genes and/or their products often work together, interacting in functional groups or pathways to contribute to phenotypic variation or susceptibility to disease. Therefore, GWASs which focus on individual genes/loci may not be optimally effective for identifying pathophysiologically significant genetic pathways underlying osteoporosis and OF. A major advantage of pathway-based GWASs is that they link the wealth of information embedded in GWAS data to our knowledge of functional biological pathways that is readily available in public databases.
Zhang et al.[44] performed the first pathway-based GWAS for osteoporosis in a population of US Caucasians, and identified the importance of the regulation-of-autophagy (ROA) pathway for ultradistal radius BMD. This is the first study implicating the ROA pathway's importance to osteoporosis, though autophagy had previously been recognized to be important in endochondral ossification.[45] Another pathway-based GWAS conducted in 700 elderly Chinese Han subjects (350 with hip OF and 350 healthy matched controls) revealed the importance of the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) pathway for hip OF.[46] In the pathway-based analyses, the maximum statistic for all SNPs near a gene was taken to represent the significance of the gene and use a permutation-based approach that shuffles the phenotypes to adjust for multiple testing.[47] Notably, most individual genes included in these significant pathways did not reach genome-wide significance in SNP-based GWASs, underscoring the importance of looking beyond the top significant SNPs/genes and searching for additional risk factors with moderate statistical significance.
4. Bivariate GWASs
Osteoporosis is highly correlated, both genetically and environmentally, with other human diseases/conditions such as obesity (characterized by high body mass index [BMI]) and sarcopenia (characterized by low lean mass and strength). GWASs, however, have typically been implemented in a univariate framework, analyzing different phenotypes (e.g., BMD or BMI) independently, and ignoring the potential genetic correlation between disease traits. Thus, this approach is not well suited for detecting pleiotropic genes, which may exist for two genetically correlated diseases. One potentially effective strategy for identifying pleiotropic genes is to analyze potentially correlated disease phenotypes simultaneously via a multivariate GWAS approach.[47] This new strategy, which utilizes GWAS data more efficiently than univariate approaches, may help identify pleiotropic genes underlying diseases of shared genetic susceptibility, thereby revealing interconnected pathophysiological networks for a spectrum of common human diseases.
The first bivariate GWAS analysis for osteoporosis and obesity, conducted in 1,000 unrelated US Caucasians, suggested the SOX6 gene's importance in co-regulation of osteoporosis and obesity.[48] This gene was not significant at the genome-wide level in separate analyses for either BMD or BMI, highlighting the power of bivariate analyses in discovering novel pleotropic genes/loci. The SOX6 gene was independently found to be significant at the genome-wide level in GEFOS-2 [12] and was suggestively significant at the genome-wide level in the study of Hsu et al.[27] Identifying genes with pleiotropic effects, such as SOX6, may offer deeper insights into the pathophysiology of osteoporosis, and potentially, may reveal key mechanistic links between osteoporosis and obesity, two major public health problems.
5. The first whole genome sequencing study
GWASs typically identify common variants important for common human disorders. Many susceptible rare variants may be missed in GWASs due to the limited power and/or resolution of genotyping.[49,50] Recent advances in next-generation sequencing technologies have greatly enhanced our ability to discover functional rare variants.[51] Styrkarsdottir et al.[9] published the first whole genome sequencing study for osteoporosis. Through whole-genome sequencing of 2,230 Icelandic individuals with discordant BMD values, they found a rare nonsense mutation within the leucine-rich repeat containing G protein-coupled receptor 4 (LGR4) gene that was strongly associated with low BMD and OF. This mutation leads to termination of LGR4 at position 126 and fully disrupts its function. A potential problem with OF analysis is that all types of fractures were grouped together, which may introduce false positive or negative results as aforementioned. The phenotype of carriers of the c.376C>T mutation overlaps that of Lgr4 mutant mice.[9] Interestingly, although this mutation was associated with a wide range of phenotypes across species (i.e., humans and mice), it was not present in Danish and Australians.[9] Therefore, its effects in other human populations need to be further evaluated and validated.
INTERPRETATION OF INCONSISTENT RESULTS
Through the identification of novel genes, CNVs, and biological pathways that are associated with osteoporosis, recent GWASs and large-scale meta-analyses have greatly advanced our understanding of the pathophysiology of osteoporosis and OF. As illustrated above, however, comparison of the results across individual GWASs and meta-analyses raises a number of critical questions.
1) Are individual GWASs still useful when meta-analysis, with a much larger sample size, is available?
2) Why did some genes/loci identified as being significant in independent individual GWASs (e.g., ADAMTS18 in Ref [30]) fail to attain significance in independent GEFOS-2 meta-analyses,[12] which had a much larger sample size?
3) Why did some genes/loci identified as being significant in individual GWASs (e.g., RAP1A, TBC1D8, and OSBPL1A in Ref [27]) fail to attain significance at the genome-wide level in GEFOS-2, in circumstances where the samples used in the individual GWASs were included in GEFOS-2 meta-analyses?[12]
4) Why have inconsistent findings even been observed between meta-analyses (e.g., GEFOS-1[11] and GEFOS-2[12] ) whose samples overlapped to a large extent?
5) Should the meta-analysis with the largest number of individuals be considered as the gold standard to evaluate findings of other independent studies (especially relatively small GWASs)?
To address these critical questions, we performed a series of theoretical analyses using simulation studies and published our results elsewhere.[52] Our theoretical analyses, under ideal situations, demonstrated that:
1) Although the power of an individual GWAS study (of average sample size) to identify any particular locus (of average effect size) is limited, the power to identify at least one (any one) locus can be high. This may explain the observation that a number of previous individual GWASs have identified novel loci despite the relatively limited sample size of each of the studies. Given the anticipated large number of significant loci that have eluded detection thus far, individual studies are still valuable in identifying at least some of these underlying effect loci.
2) The number of loci that can be detected in meta-analyses greatly exceeds the number that can be detected in individual GWASs. However, the power of a meta-analysis to identify many independent loci simultaneously can still be limited.
3) The meta-analysis has rather limited power to replicate the findings of particular loci from individual GWASs at the genome-wide significance level, particularly for SNPs with small effects, implying inconsistent findings between independent individual GWASs and meta-analysis is not unexpected.
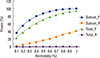
4) Adding heterogeneous samples into a subset of homogeneous samples can reduce power for a meta-analysis, rather than having the anticipated effect of increasing power due to increased sample size. This was clearly shown in our published theoretical study[52] which examined the effects of sample heterogeneity by simulating a subset of samples with true effects and the other subset of samples without effects in a meta-analysis (based on GEFOS-2). Figure (adapted from [52]) illustrates that heterogeneity results in power loss in both the mixed-effects and random-effects models. This effect was further demonstrated in our more comprehensive theoretical analyses which considered a wide range of situations and scenarios.[53] Therefore, selecting samples with homogeneous effects may be at least as important as enlarging sample size for meta-analysis. This observation may help explain the inconsistent findings between GEFOS-1 [11] and GEFOS-2 [12] despite the fact that the latter included the majority of samples in the former, and approximately 3 times as many total samples.
Our theoretical analysis was not intended to denigrate the value and importance of GWAS meta-analysis. Large-scale meta-analysis clearly represents a powerful tool for identifying novel genetic variants for osteoporosis. Nevertheless, it is critical to recognize that meta-analysis may introduce false positive/negative results due to heterogeneity and other confounding factors. Consequently, it is important to exert caution when designing studies, and interpreting results generated by meta-analysis of GWASs. In particular, the results of meta-analysis should not be used to evaluate the validity of findings from individual GWASs. Well-designed individual GWASs, using homogenous samples, have significant potential to identify additional novel genes/loci and related biological pathways for osteoporosis and OF that may not necessarily be significant in meta-analysis with larger samples, of more variable heterogeneity.
IMPLICATIONS AND PERSPECTIVES
Over the past 4 years, individual GWASs and meta-analyses of GWASs have highlighted more than 70 genes/loci and related biological pathways that contribute to the pathophysiology of osteoporosis and/or OF. Some of these genes, such as WNT, RANK-RANKL-OPG, have known functional relevance to bone metabolism and endochondral ossification, and their contribution to osteoporosis has been well established in earlier candidate gene studies.[5,54,55] The function of these established genes, proteins and related biological pathways has been reviewed elsewhere.[10] However, the purpose of GWASs is not merely to verify previous findings but, more importantly, to identify novel genes and pathways associated with complex diseases. It is notable that more than half of the genes/loci listed in Table 1 are novel, though their functional importance to bone metabolism awaits validation, ultimately through molecular functional studies.
Collectively, the genes/loci identified from individual GWASs and meta-analyses, to date, explain less than 6% of the variance in BMD variation. Therefore, further endeavors are needed to explore undiscovered genetic factors associated with BMD variation. At the DNA level, there are several possible paths towards uncovering these novel and elusive genes:
1) Well-designed, powerful individual GWASs. For such studies to be powerful, well-defined and homogeneous phenotypes measured with high data quality and accuracy, large and homogeneous samples, and comprehensive statistical and bioinformatical analyses should be needed.
2) Meta-analyses with even larger samples than GEFOS-2, which must be executed with caution because of the power loss associated with between-study heterogeneity and other confounding factors.
3) Meta-analyses with smaller sample sizes, but with less between-sample heterogeneity, to reduce the probability of generating false positive and false negative results.
4) Utilizing available GWAS data to perform additional analyses such as CNV, pathway based, and multivariate analyses.
5) Performing genetic studies focused directly on OF, rather than BMD or other less critical osteoporosis phenotypes. For OF studies, large and homogeneous samples of the same type of OF are critically important and essential. Although it is tempting to mix different types of OFs to increase sample size, heterogeneity may contribute to false findings and results that complicate data interpretation.
Current evidence suggests that the genetic architecture of osteoporosis and OF is complex, involving both common and rare functional variants.[50,51] These findings are similar for other complex human diseases that represent significant public health problems (e.g., obesity, diabetes, and cancers). GWASs and meta-analyses are largely designed to identify common variants. Thus, many susceptible rare variants may be missed in GWASs due to the limited power and/or resolution of genotyping. Recent advances in next-generation sequencing technologies, however, have greatly enhanced our ability to discover functional rare variants.[51] Unfortunately, large-scale whole genome re-sequencing studies are currently prohibitively expensive. More economically feasible approaches might involve re-sequencing of targeted genes/loci, or exome sequencing studies. Another potential approach to identify rare variants is to perform imputation analyses based on publicly available genome sequence data. The recent 1,000 Genomes Project (http://www.1000genomes.org/) produced a comprehensive catalog of human genomic variants, in particular rare variants.[56,57] Thus, through imputation of currently available GWAS samples, it is now feasible to identify/infer the majority of known human genomic variants (including rare variants) for these association studies. As technological advances decrease the unit price for genome re-sequencing, it is expected that large and powerful genome-wide re-sequencing studies will become feasible in the near future, resulting in the identification of numerous rare genetic variants that effect osteoporosis and OF. It is critical to recognize that in order to identify rare variants with reasonable power, re-sequencing studies will require very large sample sizes due to the low minor allele frequencies of these rare variants.
GWASs and re-sequencing studies focus on DNA sequence variants alone. Recent studies, however, have shown that epigenetic regulation is also critical to the pathophysiology of many human complex diseases, and may partially account for the heritability that has not been accounted for based solely on DNA variants.[58] Epigenetics refers to reversible, heritable changes in gene regulation that occur without a change in DNA sequence.[59] Consequently, in order to identify additional and novel heritable factors contributing to BMD variation and osteoporosis risk, it has become necessary and timely to study epigenomic regulation in relevant bone-related cells, including DNA methylation, histone modification, microRNA and long non-coding RNAs.
Protein post-translational modifications, such as protein phosphorylation, have also been shown to play significant roles in regulating gene expression,[60,61] signal transduction,[62,63] and cellular functions[64] closely related to bone metabolism. Functional studies at the protein level, including protein expression[28,65] and post-translational modification, may also contribute to our comprehensive identification and understanding of cellular and molecular mechanisms regulating bone metabolism.
GWASs are ultimately studies at the DNA level, and the above comments illustrate the importance of studying complex human diseases from a systems biology perspective.[66,67] Gene expression is a complex process that is regulated simultaneously and interactively at DNA, RNA, protein, epigenomic and environmental levels. Therefore, a genomic convergence or systems biology approach that organically integrates the information from GWASs, gene expression, proteomics, epigenomics, protein post-translational modification, and gene-environment studies may help facilitate the identification of key pathways that are globally involved in the pathogenesis of osteoporosis and OF. For example, using a systems genetic analytic approach, Calabrese et al. identified a physiologically relevant gene network and used it to discover novel genes and regulatory mechanisms involved in the function of osteoblast-lineage cells.[68] In another study, Deng et al. ascertained SOD2 as a susceptibility gene for osteoporosis in Chinese by integrating evidence from DNA, RNA, and protein levels.[69] Ultimately, the functional relevance of the identified variants needs to be confirmed by in vivo and/or in vitro molecular biology studies.


 XML Download
XML Download