

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The growth, repair and maintenance of the skeleton is under the control of circulating hormones, the nervous system and very many locally generated effectors that regulate its modelling and remodelling. Modelling or the construction of bone, takes place from the beginning of skeletogenesis during fetal life, until the end of the second decade when the longitudinal growth of the skeleton is completed. It is responsible for determining the size and shape of bone. During bone modelling, bone is formed and deposited on the outer surface of the bone, thus widening the lengthening bone. Simultaneous resorption of bone from its inner surface adjacent to the medullary cavity, enlarges the medullary cavity that houses the marrow cells.
Bone remodelling is carried out by osteoclasts, cells that resorb bone, and osteoblasts, cells that form bone. These are the two main effector cells of the basic multicellular units (BMUs), providing sites for the removal and replacement of damaged or old bone by new bone throughout adult life. Remodelling is also a means of adapting the skeleton to changes in loading, and is an integral part of the calcium homeostatic system.
In considering therapeutic approaches to the prevention and treatment of bone loss, it is essential to keep in mind the ways in which communications take place among cells of bone to achieve and maintain its structure. In this brief review we will summarise those mechanisms, what can be achieved with anabolic therapies for the skeleton with current approaches, and what might be hoped for in the future.
1. Cells of bone
The word "osteoblast" is often used to encompasss all members of the osteoblast lineage, and it is best to keep that in mind when reading in the subject. In remodelling, mesenchymal stem cell precursors need to differentiate through pre-osteoblast stages to mature osteoblasts, recognized histologically as plump, cuboidal, mononuclear cells residing in groups on the matrix that they have synthesized.[1,2,3]
Bone lining cells are flattened osteoblast lineage cells that are regarded as osteoblasts that have completed their synthetic function. They are much more abundant than synthesizing osteoblasts, and cover the surface components of bone, where they are thought to serve as a barrier to osteoclasts to be broached in response to need, and may be capable also of reinitiating their bone - forming activity.[4]
Osteocytes are terminally differentiated osteoblasts which have become trapped within the bone matrix behind the advancing mineralization front. They become embedded in lacunae within the bone matrix, and connect with each other and with surface cells by their intercellular processes in fluid-containing canaliculae. Osteocytes are the most abundant cell in bone (85-90%) and are very long-lived. They respond to changes in physical forces on bone and to damage, leading them to transmit signals to surface cells through canalicular processes. Among their most important protein products is sclerostin, product of the sost gene, and a powerful inhibitor of bone formation by inhibiting Wnt signalling. This will be discussed in more detail.
Osteoclasts, the only cell capable of resorbing bone, are giant multinucleated cells arising from hemopoietic precursors. They maintain an acid microcompartment under the ruffled border which they form adjacent to the bone surface,[5] forming a sealed compartment that is acidified through the active transport of protons driven by a V-type H+adenosine triphosphatase (ATPase). The passive transport of chloride through chloride channel (ClC)-7 preserves electroneutrality. This results in dissolution of bone mineral exposing the organic matrix to proteolytic enzymes, particularly cathepsin K, that degrade the organic matrix. Inactivation of any of these pathways by genetic or pharmacologic means results in failure of osteoclasts to resorb bone.[6]
The common and essential factor mediating osteoclast formation in response to all known stimuli is receptor activator of nuclear factor-kappa B ligand (RANKL) that binds to its receptor, RANK, on hemopoietic precursors to promote osteoclast differentiation as well as their survival and activity.[7] The decoy receptor, osteoprotegerin (OPG), is an essential paracrine regulator of osteoclast formation, produced by the osteoblasts and binding RANKL to prevent its promotion of osteoclast formation through its receptor, RANK. Thus regulated production of RANKL and of its local 'brake' mechanism, OPG, are essential for maintenance of normal bone remodelling.[8] This presented itself as an obvious target for anti-resorptive drug development.
2. Bone remodelling
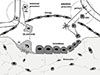
Bone remodelling is initiated asynchronously at sites that are geographically and chronologically separated from each other so that at some locations bone is being resorbed by BMUs while at others the BMUs are in their formation phase The first essential step in remodelling is the generation of active osteoclasts from hemopoietic precursors. Regardless of the source of the initiation signal, osteoclasts are derived from early and late precursors available in marrow adjacent to activation sites, or could be recruited from blood available at the bone interface through a sinus structure of bone remodelling compartments (BRCs) that have been identified and described in human bone (Fig. 1).[9,10] At each of these sites, the resorption of a volume of bone is followed by formation of new bone formation to fill the space. The BMU resorbs and replaces old bone at the same location so there is no change in bone size or shape. After a certain amount of bone is removed as a result of osteoclastic resorption and the osteoclasts have either died or moved away from the site, a reversal phase takes place in which the cement line is laid down. Osteoblasts then synthesize matrix, which becomes mineralized. Provided that equal volumes of bone are removed and replaced there will be no loss of bone or compromise in its structure during growth.
One of the sources of osteoblast and osteoclast precursors for the BRC is via the circulation, through capillaries penetrating the canopy that overlies the BRC (Fig. 1).[11,12,13] Osteoclast formation can take place rapidly in vivo, perhaps because there may be niches of partially differentiated cells available in the BRC,[14,15] having arrived there in the circulation.[16] Osteoblast progenitors are associated with vascular structures in the marrow and there may also be common progenitors giving rise to cells forming the blood vessel and pluripotent perivascular cells.[17,18,19,20,21]
The resorption activity in a BMU in adult human bone takes approximately 3 weeks and the formation response 3 to 4 months, such that remodelling replaces about 5-10% of the skeleton each year, with the entire adult human skeleton replaced in 10 years. Remodelling continues in the skeleton over the age of 50, with the purposes of repair and removal of old bone. However, as age advances less bone is deposited than was removed so remodelling of damage repair occurs.
Figure 1 summarises the interactions referred to above. An essential feature of bone remodelling that bears upon any therapeutic approach, is that within the BMU the processes of bone formation and resorption are coupled. Just as the osteoblastic lineage cells control osteoclast formation, so too the products of bone resorption and of the osteoclasts themselves, promote osteoblast differentiation from precursors in the BMU, and hence bone formation.[22,23,24,25] It is the latter communication mechanism that is referred to as "coupling".
3. Osteoporosis
In young adulthood, bone remodelling proceeds slowly, removing and replacing damaged bone with new bone. At the cellular level, the volumes of bone resorbed by the osteoclasts of a BMU and formed by the osteoblasts of that BMU are equal, so no permanent bone loss occurs. Around midlife in women, periosteal apposition virtually ceases, the volume of bone formed in the BMU by osteoblasts becomes less than that resorbed by the osteoclasts, producing a negative bone or BMU "balance". In addition, with loss of sex hormones due to ovarian failure, and in both sexes later in life, remodelling rate increases; there are more BMUs formed and each BMU removes more bone than it subsequently deposits, resulting in structural deterioration; trabeculae become thinner and less connected, cortices become thin and porous. Bone is progressively lost, resulting in bone fragility and a predisposition to fractures.
Until recently, drug treatments have been entirely focussed on prevention of resorption. These will be considered briefly, then new approaches focussed on bone forming agents will be discussed.
4. Anti-resorptives
As understanding of bone biology increased, new insights guided the development of anti-resorptive therapies for osteoporosis. The clinical outcomes of these new therapies may be predicted because the actions of the selected targets are known, and in some cases preclinical evidence fulfilled those predictions. The aims were to develop therapies to improve the fracture risk reduction if possible, to avoid the possibility of long term effects on bone structure, to find drugs whose effects reverse with cessation of therapy and drugs that inhibit resorption without inhibiting bone formation at the same time.
1) RANKL inhibition in therapy
The most commonly used treatments used clinically for osteoporosis in many countries are any of several bisphosphonates, and increasingly, Denosumab (Amgen). Studies establishing the essential physiological roles of RANKL and OPG in controlling osteoclast formation and activity, revealing a pathway obviously rich in targets for pharmaceutical development. Denosumab is a fully human monoclonal antibody that binds with high affinity and specificity to RANKL to inhibit its action. It has been revealed to have an exceptionally prolonged and powerful action, and is used by subcutaneous injection every 6 months.[26] Denosumab reduces the generation of bone remodelling units by preventing RANKL from promoting osteoclast formation and from maintaining the activity of existing osteoclasts already resorbing bone. The phase III study using subcutaneous injection of denosumab every 6 months resulted in substantial inhibition of vertebral, non-vertebral and hip fractures, and striking suppression of bone turnover that was is even more marked than with the most effective bisphosphonates.[26] Any discontinuation of treatment was associated with return of bone mineral density (BMD) to baseline, and increase again when rechallenged with treatment. Discontinuation also resulted in a rapid rise in resorption markers, indeed overshooting above control levels. This also corrected with resumed treatment.
Quite a different mechanism operates with a new class of drug, the cathepsin K inhibitors that may spare bone formation. Cathepsin K is selectively expressed in osteoclasts and discharged into the acidified sealing zone to degrade the collagenous matrix of bone. Defects in the gene encoding cathepsin K are linked to the clinical condition pycnodysostosis (OMIM 265800), an autosomal recessive dysplasia characterized by skeletal defects including dense, brittle bones, short stature and poor bone remodelling.[27] Similarly, the deletion of the cathepsin K gene in mice resulted in osteopetrosis.[28,29] The rationale of using cathepsin K inhibition is that it will inhibit the resorption of osteoclasts, without preventing them from secreting activities that can contribute to bone formation - so called coupling factor activities.[30]
2) Cathepsin K inhibition
The most advanced in development is odanacatib (MK-0822; Merck and Co., Inc, Whitehouse Station, NJ, USA), which has completed Phase II. Odanacatib is a potent, selective cathepsin K inhibitor with a long half-life (45-50 hours) that has allowed it to be used in weekly oral dosage in clinical study. In preclinical models in mouse and rabbit and in some monkey studies, cathepsin K inhibition reduced bone resorption without inhibiting bone formation.[30,31] Osteoclast numbers on bone increase, but their resorption capacity is disabled.[30] Monkey treatment studies show that odanacatib is an effective resorption inhibitor which is dose-dependent.[32,33] Interestingly there was some evidence to suggest continued periosteal bone formation. There is no obvious explanation for this and it requires confirmation.
The human Phase II study showed decreased bone resorption markers in response to odanacatib, with seemingly less decrease in bone formation markers.[34] As is the case with Denosumab, discontinuation of treatment was associated with return of BMD and markers to baseline (more rapidly than with Denosumab), and increase again with resumed treatment. At the time of writing the outcome of the large Phase III fracture study is expected in early 2014. Assuming its positive outcome, among features of great interest with this new class of compound will be how well maintained is bone formation, what will this mean for effects on bone quality, and whether combination therapy with parathyroid hormone (PTH) will be more effective with such a resorption inhibitor that does not inhibit bone formation.
5. Anabolic agents
Anti-resorptive agents do not reconstruct the skeleton, but until recently no therapeutic approach was available to restore bone once it had been lost. That situation has changed with the development of PTH as an anabolic therapy for the skeleton, despite its better known action as a resorptive hormone. The approved therapies in several countries are PTH (1-34) and PTH (1-84). Searches continue for low molecular weight peptide or even non-peptide mimics that can activate through the specific G protein-coupled receptor PTH-like hormone receptor 1 (PTHR1).
1) How PTH exerts its anabolic effect
The anabolic effectiveness of PTH requires that it be administered intermittently. In its clinical application as an anabolic therapy, PTH is administered by daily subcutaneous injection,[35] with PTH (1-34) approved for the treatment of osteoporosis in a number of countries. The pharmacokinetics required for this effect are that a peak of circulating PTH is required, returning to control levels within 3 hours. Prolongation of elevated levels brings into play the stimulation by PTH of osteoclast formation and bone resorption.[36] This resorption effect is enhanced greatly with infusion of PTH over some hours,[37] or with the consistently elevated PTH of primary hyperparathyroidism. This pharmacokinetic requirement is well illustrated by the attempts to develop anabolic therapies by using calcilytic agents to release PTH from the parathyroid gland. These attempts to achieve short-lived peaks of circulating PTH have so far not been successful.[38,39,40]
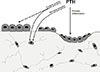
Studies of PTH pre- and post-treatment bone biopsies in women indicated that the predominant PTH effect was to increase remodelling, with some lesser effect on modelling.[41,42] Thus the anabolic effect of PTH has two components, a remodelling dependent effect said to account for over 70% and a modelling based effect accounting for the remaining 30% of the anabolic effect. Current views of the anabolic action of PTH are that it increases the recruitment and activation of BMU's, that it acts on committed osteoblast precursors to promote their differentiation, inhibits osteoblast and osteocyte apoptosis,[43] and inhibits the production of the bone formation inhibitor, sclerostin.[44] There is also much interest in the possibility that PTH treatment results in transient activation of osteoclasts, that in turn produce activity that enhances the osteoblast differentiation effect. The latter may be independent of resorption,[45,46] or may result from the release of growth factors (transforming growth factor beta [TGFβ], insulin-like growth factor-1 [IGF-1]) in the resorption process[47,48] that enhance the availability of mesenchymal stem cells (TGFβ), or their differentiation in the osteoblast lineage (IGF-1). These aspects of the anabolic action of PTH are summarised in Figure 2.
When PTH is used by daily injection to promote bone formation, increased blood levels of bone formation markers (e.g. amino-terminal propeptide [P1NP]) are detected within weeks, followed after a delay of some months by increased circulating and urine bone resorption markers. The gap between the two has been referred to as the "anabolic window", based on the thought that PTH is first anabolic through an effect on modelling, then catabolic through remodelling.[49] Such a switch has seemed unlikely, and there may be a simpler mechanistic explanation. The direct actions of PTH on osteoblast lineage cells depicted in Figure 2 is expected to result in rapid release of cell-derived P1NP. On the other hand, with each activated BMU in response to PTH there would be a gradual accumulation of resorption products as a result of osteoclast activity, culminating eventually in increased detectable circulating resorption markers. Such an explanation for the time delay between increases in formation and resorption markers explains the greater resorptive effects of higher doses of PTH, as well as the predominant resorptive effects of PTH secretagogues studied so far.
The resorption component of PTH action therefore continues to be of interest, since an effect at the BMU of as short duration as possible could minimise stimulation of osteoclast formation and activity that occurs with repeated injection of PTH (1-34) or PTH (1-84). This has been at the centre of discussion of data obtained with the trial of daily injections of PTH-related protein (PTHrP) (1-36), which has been suggested to have a purely anabolic action because increases in resorption markers are minimal.[50] Explanations offered for this are that either PTH and PTHrP (1-36) have different pharmacokinetics,[51] or that PTH (1-34) action at the receptor is more prolonged than that of PTHrP (1-36), for which there is in vitro evidence.[52] Certainly, PTHrP (1-36), like the remainder of PTHrP, is very susceptible to proteolytic degradation, and this is reflected in the fact that high doses are required in daily injections to achieve effects. In a comparative study,[53] the doses administered of PTHrP (1-36) were 20-fold higher than those of PTH (1-34). The two treatments had similar effects on BMD, the effect on bone formation marker, P1NP, was significantly greater with PTH (1-34) treatment, and the incidence of hypercalcemia was higher in those treated with PTHrP (1-36).
2) PTH and anti-resorptive therapies
There is much to be learned about the clinical use of treatment with PTH. An early lesson was that the increased bone mass benefits obtained with treatment were lost rapidly after treatment discontinuation, emphasizing the importance of following course of PTH treatment with anti-resorptives.[54,55,56] At present the favoured anti-resorptives for this purpose are bisphosphonates or anti-RANKL. It will be of considerable interest to see whether cathepsin K inhibition offers any advantage in this respect. It might theoretically be predicted to do so if its proposed mechanism of preserving a coupling effect[30] stands up to further scrutiny.
Another question of major interest is whether there is any logic, or any further benefit to be obtained, from concurrent treatment with PTH and an anti-resorptive. The thought that osteoclasts might be required for the anabolic action of PTH arose when the anabolic effect of PTH was significantly reduced in sheep co-administered a bisphosphonate (Tiludronate) as an inhibitor of bone resorption.[57] Some, but not all studies of the PTH anabolic effect in rats treated concomitantly with bisphosphonates have also shown impaired anabolic responses. The hypothesis that giving the two treatments together would be more effective than either alone was addressed in two clinical studies.[58,59] In fact the combined treatment resulted in inhibition of the response to PTH as assessed by computed tomography (CT), BMD and biochemical markers. This has been investigated further using BMD as the primary endpoint in evaluating the outcome of PTH combined with either Denosumab,[60] or with zoledronate in a 1-year study, [61] with each showing a greater effect of the combination than the anti-resorptive alone.
A problem with these studies is their reliance upon BMD measurements for assessing the combined effects. PTH and bisphosphonates produce their effects on BMD in very different ways, PTH by producing new bone tissue, which is initially under-mineralized, while alendronate maintains the same amount of bone tissue that undergoes more complete secondary mineralization because of the suppression of the bone remodelling rate. It seems inappropriate to add these two BMD values that are achieved in such different ways. Nevertheless the possible benefit of concurrent treatment with PTH and an anti-resorptive needs continued consideration and study. Some preclinical data is suggestive of such a benefit. Co-treatment with high dose PTH in mice with RANKL or bisphosphonate inhibition or genetic knockout of RANKL resulted in increases in some indices of bone formation.[62]
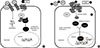
Thus the suggestion from some clinical studies and from the mouse study is that in the absence of osteoclasts, PTH can still exert an anabolic action to some extent, through any or all of three possible means: (i) direct action on osteoblast lineage cells within BMU's that were existing at the time of RANKL blockade, (ii) direct modelling action of PTH, and (iii) PTH inhibition of sclerostin production by osteocytes. This is illustrated in Figure 3. A prediction from this would be that there would be a plateau of anabolic effect reached, perhaps appreciably earlier than with PTH alone. Whether such combination treatment approaches will have favourable effects on structure or on fracture incidence will be a difficult question to answer, largely because there will be great reluctance to undertake the large clinical studies needed to assess fracture risk.
3) PTH - future use
What does the future offer in relation to PTH in therapy? It seems to have much to offer as a means of rebuilding the skeleton after bone is lost. The major clinical trial showing anti-fracture efficacy of PTH[35] was stopped at 18 months because of osteosarcoma occurrence in a prolonged toxicity study in rats,[63] resulting in a "black box" warning by the Food and Drug Administration (FDA) in the USA. There has been no evidence to suggest in human studies or in clinical use that osteosarcoma is a potential side effect of PTH treatment.[64] As a result of the FDA warning, treatment with PTH is generally limited to 18 months. That might indeed be sufficient, especially if it is followed with anti-resorptive treatment, as seems to be strongly indicated. Furthermore a case has been made for the use of intermittent, shorter course of daily PTH treatment. For example, in a group of patients after 12 months' alendronate treatment, daily PTH plus alendronate was as effective as alendronate plus three cycles of 3 months PTH with 3-month intervals, with the end-points being BMD and biochemical markers.[65]
PTH remains an expensive treatment regimen, and many health provider systems cannot afford to have it used to the extent that it might be, based on quality evidence. Indeed it is in several countries restricted to use in those patients regarded as having "severe" osteoporosis, with multiple fractures and extremely low BMD. The problem of cost might eventually be alleviated, especially if new approaches to treatment can be developed that make use of the PTH anabolic pathway. Certainly, the availability of an anabolic therapy after having to rely on anti-resorptives alone has stimulated great interest in this approach, and it is likely to be very much more widely applied than it has to the present time.
6. Wnt Signalling Targets
1) Sclerostin, the SOST gene product
Recent research has shed new light on the control of bone formation, with particular interest in the possibility of modulating the activity of components of the Wnt canonical signaling pathway to produce a net anabolic effect. Sclerostin is a secreted protein encoded by the SOST gene that inhibits bone formation by binding to the Wnt co-receptor, low density lipoprotein receptor-related protein 5 (LRP5), thereby blocking its interaction with Frizzled and inhibiting Wnt signaling in the osteoblast.[66,67] Activation of the canonical Wnt signaling pathway leads to stabilization of β-catenin in the cytoplasm through inhibition of glycogen synthase kinase (GSK)-3β-mediated phosphorylation, resulting in accumulation of cytoplasmic β-catenin followed by its translocation to the nucleus and transcriptional activation of specific gene targets (Fig. 4). Such activation in mesenchymal cells inhibits chondrocyte differentiation and promotes osteoblast activity.[68]
2) Sclerostin inhibits bone formation
The first link between Wnt signaling and human bone disease came from observations that inactivating mutations in LRP5 cause the osteoporosis-pseudoglioma syndrome (OPPG, OMIM 259770) characterized by severely decreased bone mass.[69] Conversely, a syndrome of high bone mass was found to be caused by a gain-of-function mutation of LRP5 (OMIM 601884). These genetic syndromes were reproduced with the appropriate genetic manipulations in mice.[70,71] The Wnt/β-catenin signaling pathway has a number of contributing inhibitors and activators that offer several targets that may be suitable for pharmacological intervention (Fig. 4). These include extracellular agonists and the points of interaction of antagonists, especially the secreted frizzled-related proteins (SFRPs), dickkopf (DKK) proteins and sclerostin, as well as regulation within the cell of GSK-3β, the enzyme that plays a crucial role in determining availability of β-catenin for the transcriptional effects that are essential for Wnt signalling.[72,73,74] The primary aim of these interventions (Fig. 4) is to increase Wnt/β-catenin canonical signaling in order to increase bone mass. Initial success in animal models has been reported with the inhibition of DKK-1, GSK-3 and sclerostin,[74,75,76,77] but the most advanced in preclinical and now early clinical development is blockade of the action of sclerostin by treatment with a neutralizing antibody.
Sclerostin, the protein product of the sost gene, is produced primarily by osteocytes and powerfully inhibits bone formation through inhibition of Wnt signalling, sparking great interest in roles for the osteocyte in bone modeling and remodeling. Sclerostin null mice have very high bone mass, and conversely, severe osteopenia occurs in transgenic mice overexpressing sclerostin in osteocytes.[78] Loss of function Sost mutations cause the greatly increased bone mass of sclerosteosis and van Buchem's disease.[79,80] Physiologically, rapid reductions in sclerostin could signal to limit the filling of remodeling spaces by osteoblasts, in addition to maintaining the quiescent state of lining cells on non-remodeling bone surfaces.[81]
3) Blockade of sclerostin action
It was predicted that inhibition of production or action of sclerostin resulting in enhanced Wnt canonical signaling would lead to increased bone mass. That was the basis of preclinical testing of neutralising antibody against sclerostin, first in preclinical studies. A single injection of sclerostin antibody stimulated bone formation in rats with severe bone loss, either aged males or females following following ovariectomy.[74,82] In neither case were there any significant changes in resorption parameters. In a rat hind limb immobilisation model, anti-sclerostin treatment promoted bone formation and resulted in decrease in bone resorption markers.[83]
In a phase I study of anti-sclerostin (AMG 785, Amgen)[84] healthy men and women were treated for up to 85 days with escalating doses of AMG 785. This resulted in dose-related increases in bone formation markers and a decrease in the resorption marker, serum carboxyterminal cross-linked telopeptide of type I collagen (CTX). The latter observation was unexpected, perhaps related to changes in osteoblast differentiation, with less RANKL-producing cells of the osteoblast lineage available for presentation to osteoclast precursors. In this short study, BMD increased significantly at the spine (5.3%) and hip (2.8%), with five subjects at the highest dose developing detectable antibodies, two of which were neutralizing.
A 12-month phase II randomised, placebo-controlled, multi-dose study of 410 women was carried out with the humanised monoclonal antibody (Romosozumab) recapitulated the rapid increase in BMD that had been seen in the preclinical studies.[85] A significantly greater increase in BMD was obtained with Romosozumab than with either of the comparators, alendronate or teriparatide, reaching 11.3% at the lumbar spine, 4.1% at total hip, and 3.7% at femoral neck after 12 months' treatment. The bone turnover data was remarkably different from that with any other treatment. Romosozumab treatment was associated with a transitory increase in P1NP, evident after 1 week, maximal at 1 month and declining to control levels thereafter. As had been noted in the phase I study,[84] the resorption marker, CTX, declined rapidly and remained significantly reduced throughout the 12-month treatment period. The contrast with the results of PTH treatment were remarkable, with its expected rapid and sustained increase in P1NP, with increased CTX at the 3-month assay, and sustained throughout the 12 months.
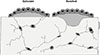
The reason for the decrease in bone formation marker after an initial rise is unknown. The decreased resorption marker might be explained by a change in the osteoblast lineage population as a result of profoundly increased bone formation, with lesser availability of cells that stimulate RANKL production and therefore osteoclasts. These aspects of anti-sclerostin action will undoubtedly be the focus of attention. They draw attention to the fact that the mechanisms involved in sclerostin blockade are new to us. In rodents and non-human primates the tissue level mechanism by which anti-sclerostin increases bone is predominantly through quiescent surfaces - thus a modelling effect. On pre-resorbed surfaces (remodelling) the new bone formed is greater than that resorbed, and includes bone laid down over quiescent surfaces adjacent to remodelling sites.[74] This is represented schematically in Figure 5.
In discussing the PTH anabolic action (above) we drew attention to the fact that cessation of treatment is followed by bone loss and increased resorption markers. That has not yet been addressed clinically with anti-sclerostin, but in a discontinuation study in ovariectomised (OVX) rats in which anti-sclerostin treatment was given for 8 weeks, then discontinued, the increase in BMD that had been achieved was maintained for a period followed by gradual decline, particularly in the lumbar vertebrae. This was associated with decline in formation and increase in resorption markers.[74] It seems likely that anti-sclerostin therapy, like that with PTH, will require the use of anti - resorptive treatment after anabolic treatment is stopped.
In the case of PTH treatment also, the question of using that anabolic therapy concurrently with an anti-resorptive has been discussed. It is a question that remains to be resolved, and the same might arise with anti-sclerostin. In accord with the fact that the latter's action is achieved predominantly through acting on quiescent surfaces, it might be not be expected that pre-treatment or combined treatment with anti-resorptive therapy would block the anabolic effect of anti-sclerostin. In a study of sequential alendronate and anti-sclerostin in OVX rats, this co-treatment did not blunt the anabolic effects of anti-sclerostin on bone formation, bone mass or bone strength.[86] Clearly this will require much further preclinical and clinical investigation. On the other hand, antisclerostin therapy is itself associated with reduced resorption, at least as assessed by circulating markers,[85] so we might well ask - do we need to consider applying concurrent ant-resorptive to anti-sclerostin therapy? Perhaps we do not.
Of particular interest is the fact that PTH rapidly reduces sclerostin mRNA and protein production by osteoblasts in vitro and in bone in vivo,[44,87] suggesting reduction of sclerostin output by osteocytes as a contributor to the anabolic effect of intermittent PTH (Fig. 2). The mechanism of this inhibition is all the more interesting with the finding[88] that the cyclic adenosine monophosphate (AMP)-mediated effect of PTH to diminish sclerostin production operates through a long range enhancer, myocyte enhancer factor 2 (MEF2), the discovery of which came from the pursuit of the nature of the van Buchem's disease mutation.[80] There may be small molecule approaches amenable to sclerostin regulation, in addition to antibody neutralization of its activity.
Any new therapy emerging from manipulation of the Wnt canonical signaling pathway will need to ensure firstly that it is safe, and secondly, that its action can be targeted specifically to bone. Wnt proteins are critical signaling proteins involved in developmental biology, with roles in early axis specification, brain patterning, intestinal development, and limb development. In adults, Wnt proteins play a vital role in tissue maintenance, with aberrations in Wnt signaling leading to diseases such as adenomatous polyposis.[72] Inhibition of GSK-3 results in increased cyclin D1, cyclin E, and c-Myc, and overexpression of these cell cycle regulators has been linked to tumour formation.[89] All relevant possibilities of side effects of enhanced Wnt signaling need to be kept in mind throughout preclinical and clinical studies.
CONCLUSION
The intercellular communication mechanisms taking place in bone have been summarized to indicate what opportunities they present for the development of drugs that build bone after it has been lost. The only currently available anabolic therapy for osteoporosis is PTH, but exciting possibilities of new anabolic therapies have been revealed through mouse and human genetics. Neutralisation of sclerostin is highly effective in preclinical studies and shows great promise clinically. No matter how effective the anabolic treatments are shown to be, however, the need for effective, safe anti-resorptives for long term use will remain, in order to preserve bone mass once it has been restored. Properly conducted clinical trials in the coming years will see the emergence of new combination therapies for bone loss that are effective, durable and safe, at an affordable cost.

 XML Download
XML Download