

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mammalian bones have two remarkable features. First, they have the unique ability to adjust their shape and size to adapt to changes in mechanical strains on the skeleton, in that they may widen or change axis of the bone by removal or addition of bone to the appropriate surfaces in response to the shift of mechanical forces. This mechanical adaptation of shaping and re-shaping of the bone structure is carried out by a process known as the bone modeling that involves independent actions of osteoblasts and osteoclasts.[1] Bone modeling occurs not only during developmental growth to allow the bone to acquire the most mechanically favorable architectural structure to provide the required bone strength for structural support and protection of internal organs.[2] Bone modeling also occurs in the adult in response to a mechanical load, such as in tennis players, in whom the radius of the playing arm has a thicker cortex and a larger periosteal circumference than the contralateral radius. The second distinctive feature is the ability of the bone to constantly "rejuvenate" or "renew" itself through a process known as the bone remodeling. Contrary to bone modeling, bone remodeling is accomplished by the collaborative and sequential actions of osteoclasts and osteoblasts to regulate bone resorption and formation in a coupled manner.[3] This process not only provides a cellular mechanism to replace damaged or fatigued bone with new, mechanically sound, bone to maintain the bone integrity but also serves to offer an access to the skeletal reservoir of essential minerals for maintenance of mineral homeostasis.
Although osteoblasts and osteoclasts, the principle cell types that mediate bone formation and resorption, respectively, are major participants of the bone modeling and remodeling process, osteocytes have key regulatory functions in both processes.[4] Osteocytes, derived from terminally differentiated osteoblasts as they become fully entrapped by matrix,[5] are the most abundant cell types in bone with a very long life span of up to 25 years.[6] A unique morphologic feature of osteocytes is that each osteocyte occupies a single lacunae and each has an extensive network of dendritic processes that extend through a series of interconnected canaliculi to make contact with other osteocytes, periosteal and endosteal lining cells, osteoclasts, and bone marrow cells.[7] This interconnected lacunae/canaliculi network provides tissue fluids and spaces for nutrition and metabolic exchange for the bone cells and also allows soluble signaling molecules and vesicles freely migrated from osteocytes to other bone cells. Thus, osteocytes are ideally positioned throughout the bone matrix to be key regulators of bone modeling and remodeling. Osteocytes also release paracrine factors, such as sclerostin (SOST)[8] and receptor activator of nuclear factor-kappa B ligand (RANKL),[9,10] to locally regulate bone formation and resorption, respectively. They can even function as an endocrine organ to secrete soluble factors (e.g., fibroblast growth factor [FGF]-23) to distant organs (e.g., the kidneys[11]) to regulate other bodily functions, such as phosphate and vitamin D homeostasis.[12,13]
The molecular mechanism by which osteocytes influence bone modeling and remodeling likely involves osteocyte-derived factors. One such potential mediator is the osteocyte-derived insulin-like growth factor (IGF)-I. Bone is one of the major target organs for IGF-I,[14,15,16] which is a potent growth factor that promotes bone formation, developmental bone growth,[17,18] and bone resorption.[19] Like other bone cells, osteocytes produce significant amounts of IGF-I.[20] While the locally-produced bone-derived IGF-I contributes little to the circulating IGF-I level, bone-derived IGF-I functions as an autocrine/paracrine effector to regulate bone turnover, and may play a more important regulatory role than the circulating liver-derived IGF-I in bone modeling and remodeling. Accordingly, global disruption of the Igf1 gene in rodents resulted in impaired longitudinal and periosteal bone expansion during postnatal growth,[18] whereas the disruption of hepatic IGF-I, which reduced the circulating IGF-I level by >75%, had no significant impact on bone growth at early life.[21] The fact that the developmental bone growth in the mutants with Igf1 conditional deletion in type 1α2 collagen-expressing osteoblasts were blunted at as early as 2 weeks of age,[22] while the conditional ablation of Igf1 from type 2α1 collagen-expressing chondrocytes did not suppress bone growth until the animals were at 8 weeks of age,[23] suggests that IGF-I derived from different bone cells may have different regulatory role in the overall bone homeostasis.
In this review, we will discuss the potential regulatory role of osteocyte-derived IGF-I in bone modeling and remodeling, by focusing on our recent findings in transgenic mice with targeted deletion of Igf1 gene in osteocytes. We will address the effects of deficient expression of osteocyte-derived IGF-I on the bone response to mechanical loading, the developmental bone growth, bone turnover response to calcium stress, and fracture repair. We will also briefly discuss the preliminary evidence that osteocyte-derived IGF-I may function as an endocrine factor to determine the size of key internal organs
1. Osteocyte-derived IGF-I is an essential mediator of mechanosensitivity in the bone
Mechanical loading is a key physiological regulatory mechanism for bone modeling.[24] While osteoblasts and other cell types are capable of sensing mechanical stimuli,[25,26,27,28] the osteocyte is believed to be the primary mechanosensory cell type in the bone. Osteocytes are strategically distributed throughout the entire bone to detect changes in loading stresses/strains; and they have the ability to communicate with and send signals to the bone surface osteoblasts and osteoclasts through the wide-spread dendritic processes connected at gap junctions.[29,30] When osteocytes sense an increase in mechanical strain as a result of loading, they send biochemical signals to the bone surface osteoblasts to stimulate bone formation and to osteoclasts to inhibit bone resorption. When the mechanical strain is reduced during unloading, osteocytes communicate with osteoclasts to initiate bone resorption to remove excessive bone. The findings that transgenic mice with ablation of osteocytes were unresponsive to unloading and had an impaired mechanotransduction[31] further exemplify the importance of osteocytes in the bone mechanosensitivity.
Previous in vitro studies have shown that the mechanotransduction mechanism in bone cells is very complex and involves a large numbers of signaling mediators and pathways.[32,33,34] Recent findings that targeted disruption of kinesin family member 3A (Kif3A),[35] polycystic kidney disease-1 (Pkd1),[36] Stat3,[37] or connexin 43[38] gene in osteocyte conditional knockout (KO) mice each markedly suppressed the bone formation response to loading in vivo indicate that mechanotransduction in osteocytes is also highly complex. There are compelling reasons to suspect that osteocyte-derived IGF-I is an important mediator of the osteogenic response to loading. Accordingly, the IGF-I signaling is a major mechanotransduction component in bone.[33] The upregulation of IGF-I expression in osteocytes is one of the earliest bone response to mechanical loading.[20,39,40] The long bones of transgenic mice with overexpression of IGF-I in bone showed enhanced osteogenic response to in vivo mechanical loading.[41] Administration of IGF-I protein stimulated bone formation only in the loaded, but not in the unloaded, bone.[42,43] Conditional disruption of Igf1 gene in type I collagen-expressing mature osteoblasts resulted in drastic loss of the osteogenic response to loading.[44] Osteocytes are descendants of mature osteoblasts. It is reasonable to assume that deletion of the Igf1 gene in mature osteoblasts would also delete the Igf1 gene in osteocytes. Thus, studies with osteoblast Igf1 conditional KO mice could not allow definitive conclusion on whether the loss of the loading-induced osteogenic response was due to deficient osteoblast-derived IGF-I or to the lack of osteocyte-derived IGF-I, or both.
To evaluate whether osteocyte-derived IGF-I functions as a key determining factor of osteogenic response to loading, we recently generated osteocyte Igf1 conditional KO mice and corresponding wild-type (WT) littermates by crossbreeding Igf1 floxed mice with Dmp1-Cre transgenic mice.[45,46] The homozygous conditional KO mice reduced the number of IGF-I-expressing osteocytes in both cortical and trabecular bones each by >50% and the bone Igf1 mRNA level by >60%. In contrast, the numbers of IGF-I-expressing osteoblasts and osteoclasts on the bone surface or IGF-I-expressing chondrocytes at the growth plate did not differ from those in corresponding WT littermates.[46] When an equivalent loading strain in the form of two-week four-point bending exercise was applied to the tibia, the loading induced a robust periosteal bone formation response in WT littermates as expected, but this loading regimen failed to elicit an osteogenic response in osteocyte Igf1 conditional KO mice.[45] The lack of an osteogenic response was not due to inadequate loads or to reduced numbers of osteocytes in the bone.[45]. Because the Igf1 expression in osteoblasts derived from these conditional KO mutants was not different from that in isolated osteoblasts of WT littermates,[45] the inability of these mutant mice to respond to loading was not due to deficient IGF-I expression in osteoblasts. Thus, we conclude that osteocyte-derived IGF-I indeed plays an important role in determining the bone mechanosensitivity.
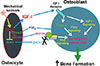
Conditional disruption of Igf1 gene in osteocytes blocked the loading-induced expression of early mechanoresponsive genes, i.e., cyclooxygenase-2 (Cox2), Igf1, and c-Fos,[45] indicating that osteocyte-derived IGF-I is a very early upstream regulator in the context of mechanotransduction.[20,39,40] The loading regimen not only did not reduce (as was seen in WT loaded bones) but in fact increased SOST expression (an inhibitor of the Wnt signaling) in osteocyte Igf1 conditional KO mutants. This, along with the findings that deficient osteocyte-derived IGF-I reduced the loading-induced upregulation of Wnt10b,[45] suggests that osteocyte-derived IGF-I is a mediator of the loading-induced activation of the canonical Wnt signaling mechanism. We have proposed a model for the molecular mechanism for the osteocyte-derived IGF-I-mediated osteogenic response to loading (Fig. 1). This model proposes that mechanical stimuli upregulate Igf1 expression in osteocytes. The osteocyte-derived IGF-I then acts as an autocrine effector to rapidly upregulate Cox2 and Wnt10b expression, and at the same time to suppress SOST expression. The secreted osteocyte-derived IGF-I and prostaglandin E2 (PGE2) in turn act on bone surface osteoblasts to activate their IGF-I signaling and PGE2 pathway, respectively. The elevated Wnt10b production and the reduced release of sclerostin then activate the canonical Wnt signaling in osteoblasts. The collective consequence of these actions leads to a synergistic increase in bone formation. Thus, osteocyte-derived IGF-I may be an integral component of the circuitry for normal loading activation response in osteoblasts.
2. Osteocyte-derived IGF-I is a key regulator of developmental bone growth
Mammalian bones continue to grow in length and width from birth until adulthood. Longitudinal bone growth takes place at the growth plate and is carried out by a process known as endochondral ossification.[47] In contrast, the bone width (size) increases at the periosteum through a process called periosteal expansion, which significantly increases bone strength, independently of increases in areal bone mineral density (BMD).[48,49] The longitudinal bone growth and periosteal expansion together are responsible for the large increase in bone mass during pre-pubertal and pubertal growth.[50,51] Developmental bone growth is governed by a complex interplay among various systemic hormones, local growth factors, nutritional factors, and mechanical factors.[47,52,53] It is widely accepted that IGF-I plays a critically important role in developmental bone growth.[53,54,55] Since conditional deletion of Igf1 gene in hepatic cells, which reduced circulating IGF-I levels by >75%, had no effects on bone length and size,[21] but targeted disruption of Igf1 gene in mature osteoblasts[22] or chondrocytes[23] greatly reduced bone length and size without affecting the circulating IGF-I level, it appears that locally produced bone-derived IGF-I, and not the circulating liver-derived IGF-I, is essential for the developmental bone growth.
Osteocyte Igf1 conditional KO mice displayed a bone phenotype of shorter bones associated with smaller periosteal circumference and cross-sectional bone area,[46] implying that osteocyte-derived IGF-I has an important regulatory role in developmental bone growth. Transgenic mice with conditional disruption of Igf1 in type I collagen-expressing mature osteoblasts[22] or in type II collagen-expressing chondrocytes[23] also exhibited a phenotype of small bone and size, but there are significant differences between osteocyte Igf1 conditional KO mutants and osteoblasts or chondrocyte Igf1 conditional KO mutants. For example, the reduced bone size in osteocyte Igf1 conditional KO mutants was associated with decreases in periosteal bone formation rate (BFR) and mineral appositional rate (MAR),[46] whereas the periosteal or endosteal BFR and MAR were not affected in chondrocyte Igf1 conditional KO mutants.[23] Osteoblast Igf1 conditional KO mice had lower BMD, but the BMD in osteocyte conditional KO mutants was not altered. Thus, osteocyte-derived IGF-I may have distinct functions than osteoblast- or chondrocyte-derived IGF-I in developmental bone growth.
The mechanism by which osteocyte-derived IGF-I influences developmental bone growth remains speculative. Both bone formation and resorption parameters in osteocyte Igf1 conditional KO mice were reduced, but each to a similar extent.[46] Thus, it appears that the impairment in developmental bone growth was associated with reduced bone turnover. Previous studies have shown that osteocytes regulate function and apoptosis of osteoblasts and osteoclasts partly through cell-cell contact or communication at gap junctions via widespread dendritic cellular processes.[29,30] Thus, it is conceivable that inadequate IGF-I expression in osteocytes could disrupt these cell-cell communication, resulting in reduced bone turnover. However, although this possibility could account for the impaired periosteal bone expansion, it does not explain why osteocyte Igf1 conditional KO mice would impair longitudinal bone growth, since growth plate chondrocytes are not in close proximity to osteocytes.[5] We speculate that the osteocytes-derived IGF-I-dependent regulation of longitudinal bone growth may involve osteocyte-derived soluble factors. A potential candidate is the IGF binding proteins (IGFBPs). IGF-I has paracrine effects on bone cell production of IGFBPs,[56] and many IGFBPs have IGF-dependent and -independent actions on bone turnover.[56,57] Changes in Igf1 expression in a number of cell types have been associated with alterations in the IGFBPs expression profile. For example, target disruption of Igf1 in chondrocytes reduced IGFBP5 expression in the growth plate cartilage.[23] Conditional disruption of Igf1 in mature osteoblasts decreased bone levels of IGFBP3 and IGFBP4. Conversely, conditional disruption of Igf1 in osteocytes increased plasma IGFBP3 level and decreased plasma IGFBP5 level,[46] raising the intriguing possibility that the reduced bone production of the stimulatory IGFBP5 and the increased bone production of the inhibitory IGFBP3 in osteocyte conditional KO mutants may in part contribute to the reduced longitudinal bone growth. Alternatively, because loading enhances longitudinal bone growth[58] and the IGF-I signaling is essential for the loading-induced osteogenic response,[33,44] the blunted longitudinal bone growth may be an indirect consequence of reduced bone mechanosensitivity.[45]
3. Osteocyte-derived IGF-I has marginal effects on the bone remodeling response to low calcium stress
There is increasing evidence that osteocytes contribute significantly to regulation of bone mineral homeostasis. Accordingly, the osteocyte is a major source of sclerostin,[8] which is a key inhibitor of the Wnt[59] and the bone morphogenetic protein (BMP) signaling.[8] The osteocyte is also a regulator of osteoclast maturation and function through modulation in osteocyte production of RANKL[9,10] and/or osteoprotegerin (OPG).[60] The osteocyte production of RANKL and sclerostin was each upregulated during calcium stress,[61,62] resulting in an increase in bone resorption and an inhibition of bone formation, respectively. Under conditions of severe calcium deficiency or an excess parathyroid function, osteocytes can also act directly to resorb bones around their lacunae and canaliculi through a process known as osteocytic remodeling.[63] This controversial concept[64,65] is supported by recent findings that 1) lactation was associated with large increases in lacunar area in tibiae and lumbar vertebrae, 2) these increases were reversed after 7 days of cessation of lactation[66], 3) this reversible lacunar remodeling was mimicked by parathyroid hormone-related protein (PTHrP)[66] or parathyroid hormone (PTH),[67,68] and 4) it was abolished by targeted deletion of type I PTH receptor in osteocytes.[66] The osteocytic remodeling is mediated by the sclerostin-mediated upregulation of carbonic anhydrase 2 in osteocytes[69] in a RANKL signaling-dependent fashion.[60] There is evidence that the osteocyte also functions as secretory cells to secrete FGF-23 to act on the kidneys to regulate phosphate and vitamin D metabolism.[11,12,13]
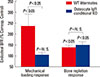
Our motivation to investigate the role of osteocyte-derived IGF-I in the osteocyte regulation of bone remodeling was based on the facts that IGF-I is a critical mediator of the skeletal response to PTH in bone cells[70] and that the PTH/PTHrP receptor signaling in the osteocyte regulation of periosteal bone formation and intracortical remodeling.[71,72] Accordingly, we have recently performed a preliminary study to assess the effects of targeted disruption of Igf1 in osteocytes on the bone and mineral homeostasis in response to the dietary calcium challenge.[73] In this study, four-week-old female osteocyte Igf1 conditional KO mice and WT littermates were subjected to a diet with low calcium content (<0.01%) for two weeks, followed by one week of normal (1.2%) calcium diet. After the dietary calcium depletion, both WT littermates and conditional KO mutants experienced severe calcium stress (reflected by a marked elevation in the plasma PTH level and reduction in the plasma calcium level). The lack of osteocyte-derived IGF-I did not affect the severity of the calcium stress. Both mouse strains lost significant amounts of bone mass and increased the number of osteoclasts along the bone surface, but there were no significant mouse strain-related differences. Although the relative increase in bone resorption (as reflected by an increase in plasma CTx level) in the conditional KO mutants was slightly greater than that in WT littermates, two-way ANOVA revealed that the effect due to deficient osteocyte Igf1 expression was only marginal (P=0.064). After one week of dietary calcium repletion, both WT littermates and conditional KO mutants showed an increase in endosteal bone formation attended by a sharp reduction in osteoclast number on bone surface. Consequently, osteocyte-derived IGF-I does not appear to be essential in the osteocyte regulation of bone and calcium homeostasis in response to dietary calcium stress. In addition, because osteocyte Igf1 conditional KO mutants showed a normal bone and calcium responses to calcium depletion and repletion, despite a complete lack of osteogenic response to mechanical loading,[45] it implies that there is a dichotomy between the mechanisms involved in bone formation responses to loading and the bone response to the dietary calcium repletion following a low dietary calcium challenge (Fig. 2). Accordingly, while osteocyte-derived IGF-I is an important mediator of mechanosensitivity of the bone, this osteocytic growth factor only has marginal effect on the osteocyte regulation of bone repletion response to a low calcium challenge. The mechanistic reason(s) contributing to the lack of a significant role in osteocyte-derived IGF-I-mediated bone repletion after a severe calcium stress is unclear. However, this severe calcium stress led to development of hyperparathyroidism and high levels of circulating 1,25-dihydroxy-vitamin D3 (1,25[OH]2D3). Both of these systemic hormones are potent stimulators of bone turnover. We speculate that the local effect of deficient osteocyte-derived IGF-I on bone turnover was probably masked by the huge systematic effects of hyperparathyroidism on bone turnover and bone loss. In any case, it is possible that while osteocyte-derived IGF-I is a potent local regulator of bone formation and resorption, it does not participate in the mechanism of systemic hormones to stimulate bone turnover. Nevertheless, much additional work is needed to resolve this intriguing discrepancy.
4. Osteocyte-derived IGF-I may function as a potential negative regulator of bone fracture repair
Recent evidence indicates that osteocyte-derived factors, particularly sclerostin, have important modulating roles in fracture healing. Accordingly, targeted deletion of the SOST gene in osteocytes has shown to enhance bone callus formation and fracture healing.[74,75] The expression of SOST gene in the fractured bone was down-regulated during the early phase of fracture healing,[76] and systemic administration of anti-sclerostin monoclonal antibody promoted fracture healing.[77,78] There are three reasons to speculate that osteocyte-derived IGF-I may have a mediating role in fracture repair. First, because IGF-I is a critical mediator of the skeletal response to PTH,[71] and PTH has been shown to promote fracture healing.[79,80] Second, IGF-I treatment has been shown to promote fracture healing.[81,82] Third and importantly, mechanical loading is believed to have beneficial effects on fracture repair,[83,84] and osteocyte-derived IGF-I is essential in determining the bone mechanosensitivity.[45]
To assess the potential role of osteocyte-derived IGF-I in fracture repair, we have performed a preliminary study to determine the effect of deficient Igf1 expression in osteocytes on the healing of tibial fractures.[85] In this study, single tibial fractures were created on 10 weeks-old male osteocyte Igf1 conditional KO and WT mice by three-point bending, and fracture healing was monitored by micro-computed tomography (µ-CT), peripheral quantitative CT (P-QCT), and histology examination. At 21 and 28 days of healing the fracture callus in each conditional KO mice exhibited significant increases in the callus bone volume fraction and trabecular number. µ-CT analysis suggested that bony union of the fracture gap appeared to be improved in the osteocyte Igf1 conditional KO fractures.[86] Histology
showed that the conditional KO fracture produced a fracture callus with more trabecular-like bone, an increase that was not due to an increase in trabecular thickness or a different basal bone formation. There was also a 85% decrease in % cartilage and >2 fold increase in the number of osteoclasts within the conditional KO callus, suggesting that the endochondral bone in the conditional KO callus had been remodeled more rapidly than WT callus, resulting in an accelerated bony bridging of the fracture gap. The mRNA levels of the bone formation genes (e.g., cbfa1, BMP2, and osteocalcin) in the conditional KO callus were also increased more than those in WT calluses.[85] These results, together, suggest that targeted deletion of Igf1 in osteocytes would result in an increase in bone callus remodeling with a balance shifted toward bone formation; the end result being an accelerated fracture healing including bridging. This preliminary study, which indicates that targeted deletion of Igf1 in osteocytes not only did not impede but in fact promoted fracture repair is unexpected and suggests that osteocyte-derived IGF-I may have a novel inhibitory function in fracture repair. The molecular mechanism by which osteocyte-derived IGF-I regulates negatively bone fracture healing is unclear and needs much further investigations. Nevertheless, because osteocyte Igf1 conditional KO mice were unresponsive to mechanical loading with an increase in bone formation,[45] the apparent enhanced fracture repair in the absence of loading osteogenic response in osteocyte Igf1 conditional KO mutants challenges the generally accepted dogma that loading is essential for an optimal fracture healing.
5. Potential role of osteocyte-derived IGF-I in the determination of internal organ size
The osteocyte Igf1 conditional KO mice, in addition to 8-12% shorter bone length and small bone size, have a corresponding 9-20% decrease in body weight and body size in as early as 2 weeks of age. The reduction in body weight and size was sustained throughout the entire growth period (Fig. 3). Because a major function for the skeleton is to provide space and strength to house and protect internal organs, it would make physiological sense that the size of internal organs in these osteocyte Igf1 conditional KO mice are also reduced in relative proportion in scale. Thus, we evaluated the effect of deficient osteocyte-derived IGF-I expression on the size of several key internal organs, such as the kidney, liver and spleen,[87] and found that the weight of these organs (Fig. 4) was also correspondingly reduced by 14-20% compared to those of WT littermates. The organ (e.g., kidneys) weight correlated strongly and positively with the body weight, bone size and length of the animals (Fig. 5). The apparent effect of osteocyte-derived IGF-I on the size of internal organs is exciting and suggests that the osteocyte and/or osteocyte-derived IGF-I may have regulatory endocrine actions on the development growth of internal organs. There is increasing evidence that the bone as well as osteocytes may function as an endocrine organ to influence the functions of other key organs, since it has been reported that the bone synthesizes and releases into circulation under-carboxylated osteocalcin, which migrates to the pancreas to increase the number and activity of pancreatic β-cells to produce insulin to improve glucose tolerance and fat metabolism.[88] More importantly, the development of a functional organ requires not only patterning mechanisms that confer proper identities for its constituent cells, but also growth-regulatory mechanisms that specify the final size of the organ. Precise control of organ size is crucial for the animal development and organ regeneration. Consequently, this preliminary study not only solidifies the concept that osteocytes are an endocrine organ but also provides insights into a potential mechanism of the osteocyte to accomplish the coordinated growth of the skeleton with key internal organs during developmental growth. It also suggests that osteocyte-derived IGF-I may play a regulatory role in the osteocyte-mediated coordinated growth of bone and internal organs.
When contemplating body growth in organ growth, one immediately considers the growth hormone (GH)/IGF-I axis. According to this concept, a system in which GH produces IGF-I, IGF-I has receptors throughout all nucleated cells in the body. This would seem to explain how there is safe-guard against in continuity of organ size in the developing mammals. This model system would tend to explain why. For example, the heart would not grow too large for the bony chest cavity. However, as mentioned above circulating plasma IGF-I does not appear to account for all changes in body growth. Moreover, organ growth in our preliminary study was reduced without any reduction in plasma IGF-I.[46] Because serum IGF-I level is a surrogate marker of circulating GH level, this raises the possibility that the skeleton has a mechanism, in addition to the GH/IGF-I axis, to determine the growth rate of internal organ size and thereby prevents incongruently in organ size compared to skeletal size, which functions to protect internal organs. The analogy would be that the system will not function properly if the protective glove is too small for the corresponding hand. This raises the possibility that there are other mechanisms that fine-tune the coordination between the protective skeleton and soft tissue organs.
Coordinated growth throughout the body of the various organ system in such an important process that more than one mechanism would seem to be required in order to ensure appropriate coordination. Several core signaling pathways, including the Notch, Wnt, transforming growth factor (TGF)β, Hedgehog, receptor protein-tyrosine kinases, nuclear receptors, and Jak/State pathways,[89] have been suggested to mediate the majority of cell fate decision. However, in reviewing the biology of organ size control, one immediately encounters the Hippo (Hpo) pathway. The Hpo signaling pathway is highly conserved from Drosophila to humans and is a major molecular pathway that determines the organ size.[90] The Hpo signaling pathway is a potent mechanism that restricts organ size by limiting cell proliferation and promoting apoptosis. At the core of this signaling mechanism in Drosophila is a protein kinase cascade consisting of protein kinases: Hpo and Warts (Wts) and their scaffold proteins: Salvador (Sal) and Mats. In this pathway, the Hpo-Sav complex phosphorylates and activates Wts-Mats complex, which in turn phosphorylates and inactivates the pro-growth transcriptional coactivator Yorkie (Yki). The 14.3.3 proteins sequester phosphorylated Yki, and prevent nuclear translocation of Yki, wherein it acts as a coactivator for the transcription enhancer factor (TEAD/TEF) family transcription factor Scalloped (Sd) and induces expression of growth-promoting genes. In humans, the homologs of Hpo, mammalian Ste20-like (MST)1/2, phosphorylate the Wts homologs, large tumor suppressor (LATS)1/2, leading to the phosphorylation of the Yki homologs, YAP and TAZ. The Hpo signaling in organ size is studied extensively in the liver.[91,92,93] The Hpo pathway is regulated by cell polarity, cell adhesion and cell junction proteins.[94] Therefore, it seems tenable that osteocyte-derived IGF-I somehow communicates with soft tissue organs to regulate the local Hpo pathway. Whatever the mechanism, we believe that it is crucial that bone (or the osteocyte) has a mechanism to establish the size of other organs, so that it can appropriately accomplish its protective function.
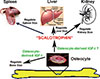
We have advanced an interesting, albeit highly speculative, mechanistic model by which osteocytes elaborate endocrine regulation of internal organ size (Fig. 6). We postulate that osteocyte-derived IGF-I is an important local regulator of the bone size; the evidence for which has been summarized above. We further speculate that osteocytes secrete a putative regulatory hormone (which we for the purpose of convenience called "scalotrophin") in relative proportion to the bone size in osteocyte-derived IGF-I-dependent manner. This putative "scalotrophin" will then serve as an endocrine effector to travel to distant internal organs to restrict their size during developmental growth. We speculate that "scalotrophin" controls the organ size primarily through modulation of the Hpo signaling. It should be emphasized that there is currently no evidence for the existence of this "scalotrophin". Nevertheless, this very interesting model merits further investigations.
CONCLUSION
Emerging evidence has indicated that osteocytes are multifunctional cells that help coordinate the various aspects of local regulation of bone metabolism and systemic regulation of phosphate and vitamin D metabolism. These regulatory functions of osteocytes are mediated in part through several key osteocyte-derived factors, including sclerostin,[8] RANKL,[9] OPG,[10] and FGF-23.[12] In this regard, IGF-I is a potent bone growth factor that regulates bone formation and bone turnover and osteocytes have been shown to express and secrete a large amounts of IGF-I, but our understanding of the functional role of osteocyte-derived IGF-I in bone and mineral metabolism is limited. As summarized in this review, our recent studies on transgenic mice with conditional disruption of the Igf1 gene in osteocytes have provided strong evidence that the osteocyte-derived IGF-I may have important regulatory roles in various aspects of bone and mineral metabolism. On the one hand, some of the suggested regulatory functions of the osteocyte-derived IGF-I are predictable. For example, the findings that osteocyte-derived IGF-I is a critically important regulator of bone mechanosensitivity is not surprising, since the osteoblast-derived IGF-I has been shown to be an essential regulator of the bone formation response to loading.[44] In addition, the concept that the osteocyte-derived IGF-I is an important regulatory of developmental bone growth is also not unexpected, since recent studies have demonstrated that locally produced osteoblast-derived IGF-I[22] or chondrocyte-derived IGF-I [23] are important regulators of the developmental bone growth. On the other hand, several findings of our studies relevant to the potential functions of osteocyte-derived IGF-I are surprising and thought provoking. First, because the bone-derived IGF-I is a potent bone growth factor that stimulates bone formation and bone turnover,[14,15,16,17,18,19] and because osteocyte-derived IGF-I appears to have an essential regulatory role in the bone formation and resorption during developmental bone growth,[46] the finding that conditional disruption of the Igf1 gene in osteocytes had only marginal effects on the bone response to depletion/repletion[73] is a big surprise. However, because the bone turnover response to dietary calcium depletion/repletion is largely regulated by systemic hormones, such as PTH and 1,25(OH)2D3, the lack of a large effect on the bone response to dietary calcium depletion/repletion in osteocyte Igf1 conditional KO mice may suggest that osteocyte-derived IGF-I is important for local regulation, but not for systemic regulation, of bone and mineral metabolism. If this conclusion is confirmed by subsequent studies, it may imply that osteocyte-derived IGF-I is a local, rather than systemic, modulator. Second, deficient expression of IGF-I in osteocytes not only did not impair, but appeared to enhance the bone regeneration process of fracture healing. These findings raises a novel concept that osteocyte-derived IGF-I functions as a potent negative modulator of endochondral bone formation in the fracture repair. This concept is novel and also thought provoking. IGF-I is a well-known bone growth factor that stimulates bone formation, and IGF-I-based therapies[95] have been suggested to have promote fracture repair. Consequently, a good understanding of the molecular mechanism by which osteocyte-derived IGF-I acts to impede fracture repair would not only help to define a functional role of osteocyte-derived IGF-I in fracture repair but could also provide a novel target for further development of novel therapeutic agents for fracture repair. Finally, the more intriguing observation is the findings that deficient in osteocyte-derived IGF-I decreased not only the bone size but also the size of key internal organs proportionally. The mechanism to limit organ size is one of the most fundamental questions in developmental biology. Accordingly, understanding of the mechanism by which osteocyte-derived IGF-I helps to determine the organ size would not only provide answers to this very important developmental question but may also lead to development of innovative concepts to promote regeneration of internal organs. In summary, osteocyte-derived IGF-I is a novel and important mediator of osteocyte regulation of bone and mineral metabolism. Further investigations of this osteocytic growth factor are warranted.



 XML Download
XML Download